

EU MDR Device Classification Guide – CE Mark Rules
For the manufacturers trying to figure out ways to place their medical devices in the EU market, classifying their devices based on their intended purpose should always be the first step. EU Classification of Medical Devices plays a key role in determining the regulatory path, conformity assessment process, and overall compliance under the Medical Devices Regulation (MDR).
As per the EU regulations, the classification is based on the risks involved in the device. It is very important to classify your device during its developmental phase as the device in each risk class has to take a different path for market authorization and the regulatory requirements differ for each class. Also, the device with higher risks has more stringent regulatory and testing requirements.
Manufacturers need to assure that the intended purpose of the device is well specified to determine the class of the device.
For easy identification of the applied rule, medical devices are categorized into Invasive, Non-Invasive, Active Devices, etc. The detailed rules for classification are specified in Annex VIII of EU MDR 2017/745.
Elexes has classified 200+ devices under EU MDR, streamlining validation and CE strategy for global clients. RA Directors and R&D heads: correct classification sets the stage for CE entry, don’t let misclassification delay market launch or increase NB costs.
Following are the various classification rules and their applicability:
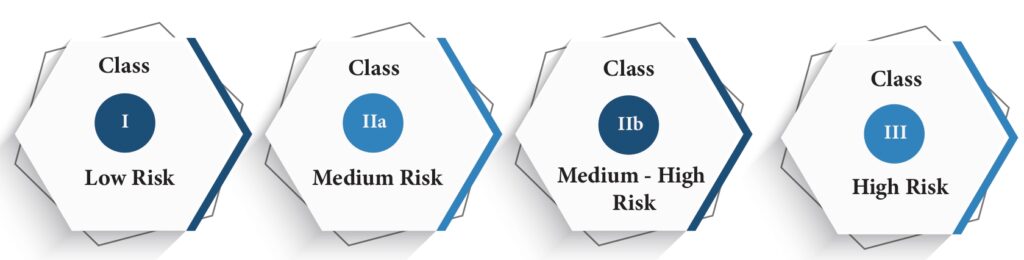
According to the above rules, the devices are to be classified in one of the following classes. This classification helps identify the conformity assessment procedure needed for the device to obtain CE marking. Also, while classifying, if a device falls under more than one class, the highest risk class has to be considered.
Manufacturers of Medical Devices who are looking to get CE Mark can contact us at jennifer@elexes.com to obtain assistance in identifying the class of your device and to decode the requirements of EU MDR 2017/745.
#elexes #healthcare #medicaldevices #cemark #eu #mdr #regulations #mdd
Download EU Classification Checklist
Book a Classification Audit with Elexes
Schedule a CE Mark Strategy Call
FAQs
What are the EU MDR Annex VIII rules?
Annex VIII of the EU MDR defines classification rules for medical devices based on invasiveness, duration of use, and part of the body affected. There are 22 rules that determine whether a device falls into Class I, IIa, IIb, or III.
How do I classify medical device software under EU MDR?
Software intended for medical purposes is typically classified under Rule 11. It is often at least Class IIa if it provides decision support, diagnosis, or therapeutic actions.
When is a Notified Body required in EU MDR?
A Notified Body is required for all device classes except Class I (non-sterile, non-measuring, non-reusable surgical). Higher classes require greater NB scrutiny during conformity assessment.
What is the difference between an accessory and a medical device?
An accessory is a product that is not a medical device on its own but is intended to enable or assist a medical device. Accessories must be classified separately based on their role.
What are Annex XVI products?
Annex XVI lists products without a medical purpose (e.g. liposuction devices, lasers for aesthetic treatment) that are still regulated under MDR due to potential risk.



















