

FDA Finalizes Quality Management System Regulation (QMSR): What Medical Device Companies Must Do Next
The U.S. FDA has formally transitioned from the legacy Quality System Regulation (21 CFR Part 820) to the Quality Management System Regulation (QMSR), marking one of the most significant regulatory shifts for medical device manufacturers in decades. The new framework establishes U.S. requirements that meet ISO 13485 standards while creating a risk-based quality management system for medical devices that operates throughout product development and manufacturing.
The QMSR implementation process for U.S. manufacturers creates a new compliance requirement that affects their readiness for inspections, their documentation systems, and their international quality control operations.
What Is QMSR and Why Does It Matter Now?
The QMSR replaces the FDA’s traditional QSR by incorporating ISO 13485:2016 by reference, while retaining specific FDA statutory requirements. The transition establishes matching requirements between U.S. manufacturing standards and international manufacturing standards while enabling the FDA to use its enforcement authority under current good manufacturing practice rules.
The QMSR framework establishes system performance requirements that FDA must use to assess regulations, while it also brings increased attention to the risk management practices, the postmarket tracking systems, and product design links to production processes. The FDA has begun enforcement of QMSR regulations, effective February 2, 2026. From that point forward, inspections will assess compliance against ISO 13485 requirements as embedded within FDA quality system regulations.
QMSR vs. Legacy QSR: What Has Changed
Key shifts introduced by QMSR include:
⦿ Direct integration of ISO 13485 clauses into U.S. regulatory compliance
⦿ Expanded expectations for risk management across the full product lifecycle
⦿ Stronger linkage between design controls, postmarket surveillance, and CAPA
⦿ Continued FDA-specific requirements for complaint handling, records access, and labeling controls
Although many manufacturing companies in the world maintain ISO 13485 certification, their compliance with QMSR requirements remains unguaranteed. The FDA will assess ISO-based systems through inspections to determine their effectiveness in meeting U.S. regulatory goals and to verify their certification status.
Who Is Impacted by QMSR?
QMSR applies broadly to:
⦿ U.S. and non-U.S. medical device manufacturers marketing in the United States
⦿ Companies operating under Investigational Device Exemption (IDE) with clinical-stage products
⦿ Contract manufacturers and specification developers
⦿ Startups transitioning from development to commercialization
Organizations that use QSR-only systems that have been in operation for a long time face the biggest threat of transition failure because their documentation systems and risk management files do not comply with ISO 13485 requirements.
Practical Next Steps for Medical Device Manufacturers
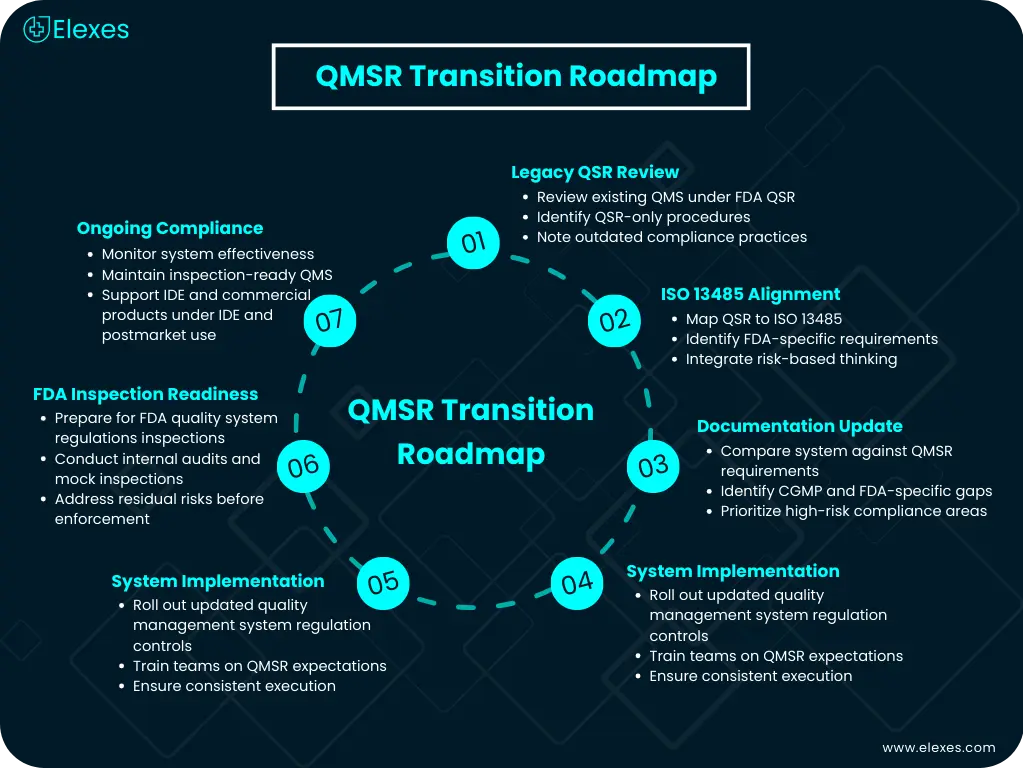
The medical device manufacturers need to take specific actions that provide practical solutions to their challenges:
⦿ Conduct a QMSR-focused gap assessment comparing existing systems against ISO 13485 and FDA-retained requirements
⦿ Update quality documentation, including quality manuals, SOPs, risk management files, and design history files
⦿ Align postmarket and CAPA processes with lifecycle risk and feedback expectations
⦿ Prepare for FDA inspections by training teams on QMSR inspection logic and evidence presentation
Early action reduces regulatory disruption, inspection findings, and costly remediation under enforcement timelines.
Why QMSR Reinforces the Need for Expert RA/QA Support

QMSR needs more than documentation because it needs officials to understand the regulations, predict inspection needs, and connect different system components. At Elexes, we view QMSR as an opportunity for manufacturers to strengthen compliance maturity while avoiding transition pitfalls.
Our QMSR services begin with gap assessments and follow with remediation planning and FDA inspection readiness to provide organizations with a compliant status and confidence before enforcement begins.
Click here to read more about the official news.
FAQs
Is ISO 13485 certification enough to comply with QMSR?
No, because ISO 13485 provides the basic requirements for QMSR, but FDA-specific requirements must be met and shown to inspectors.
When will the FDA start enforcing QMSR?
The FDA began enforcing QMSR on February 2, 2026, which follows the end of the transition period.
Does QMSR apply to investigational devices under IDE?
Yes. Manufacturers operating under an Investigational Device Exemption must maintain compliant quality systems appropriate to their development stage.
Will FDA inspections change under QMSR?
Yes, the manufacturers must maintain their quality systems in accordance with their development stage once they operate under an Investigational Device Exemption.
What is the biggest risk of delaying QMSR preparation?
Delayed preparation increases the likelihood of inspection findings, remediation costs, and potential delays to commercialization or postmarket activities.



















