

When Your Product Must Be Included in the Australian Register of Therapeutic Goods (ARTG) for TGA Medical Device Supply
Introduction
Before a product can be legally supplied in Australia, manufacturers must verify whether their product meets the regulatory definition of a medical device and whether it must be included in the Australian Register of Therapeutic Goods (ARTG) before supply in Australia.
According to guidance from the Therapeutic Goods Administration, the regulator responsible for medical devices in Australia, manufacturers must verify whether their product meets the regulatory definition of a medical device and whether it must undergo TGA medical device registration before entering the Australian market.
This determination is often more complex than it appears. Products incorporating software, AI, wellness features, or borderline clinical functionality are often misunderstood in the early stages of regulatory strategy. As a result, there can be a lack of clarity regarding medical device registration requirements in Australia and ARTG inclusion obligations.
Why Confirming ARTG Inclusion Matters
Determining whether a product requires inclusion in the Australian Register of Therapeutic Goods (ARTG) is one of the earliest regulatory decisions manufacturers must make when planning Australian market entry.
If a product that meets the definition of a medical device in Australia is not registered properly, it can lead to regulatory enforcement actions, market withdrawal, or delays in commercialization.
The Therapeutic Goods Administration expects manufacturers to assess whether their product:
⦿ Meets the legal definition of a medical device
⦿ Requires TGA medical device classification based on risk
⦿ Must undergo TGA medical device registration before supply
⦿ Falls within a regulated therapeutic goods category
For many manufacturers, particularly those in the developing software-driven or AI-enabled technologies, the question of whether their product requires ARTG registration in Australia can be a critical regulatory checkpoint early in product development.
How to Determine if Your Product Requires ARTG Registration in Australia
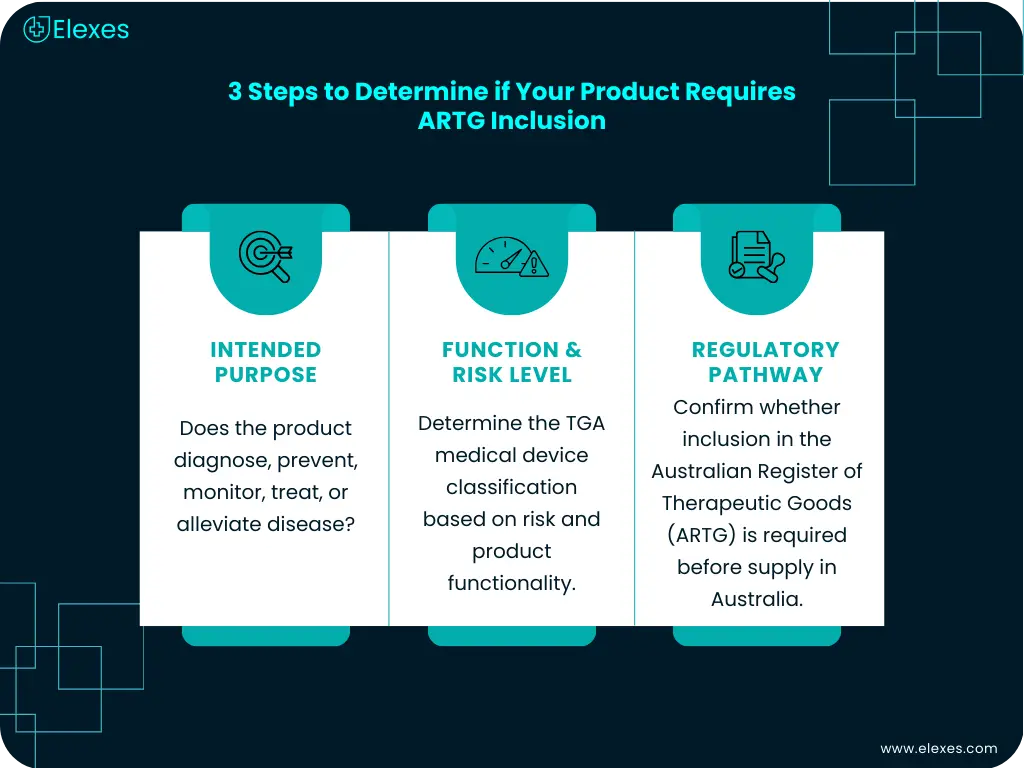
The Therapeutic Goods Administration provides guidance to help manufacturers determine whether their product must be included in the Australian Register of Therapeutic Goods (ARTG) before medical device supply is permitted.
The determination generally involves assessing three key factors:
1. Intended Purpose of the Product
The regulatory status of a product is primarily defined by its intended medical purpose. Products intended for diagnosis, prevention, monitoring, treatment, or alleviation of disease may qualify as medical devices.
2. Product Functionality and Risk
Once a product is determined to be a medical device, manufacturers must evaluate its risk level through TGA medical device classification. Classification determines the regulatory pathway and documentation required for medical device registration in Australia.
3. Regulatory Exemptions or Special Categories
Some products may fall into exemption categories or alternative regulatory pathways, while others must undergo full TGA medical device registration before supply.
Manufacturers seeking clarity often begin by asking “How to register a medical device” in Australia, but the more important first step is confirming whether registration is required at all.
The Risks of Getting the Decision Wrong

Incorrectly determining whether a product requires inclusion in the Australian Register of Therapeutic Goods (ARTG) can have significant regulatory and commercial consequences.
Potential risks include:
⦿ Delays in Australian market entry
⦿ Regulatory compliance actions from the Therapeutic Goods Administration (TGA)
⦿ Product supply restrictions or recalls
⦿ Additional regulatory remediation costs
⦿ Loss of investor or partner confidence
These risks are especially common in emerging product categories such as digital health platforms, AI-driven diagnostics, and software-enabled monitoring tools where TGA medical device classification may not be immediately obvious.
How Elexes Supports ARTG Eligibility and Market Entry Strategy
Determining whether a product requires inclusion in the Australian Register of Therapeutic Goods (ARTG) is not simply an administrative step, it is a strategic regulatory decision.
Elexes helps manufacturers determine ARTG eligibility and build regulatory strategies for medical device supply in Australia. Our support includes:
⦿ Medical device regulatory strategy for Australia
⦿ TGA medical device classification assessments
⦿ Determining medical device registration requirements in Australia
⦿ Preparing documentation for TGA medical device registration
⦿ Regulatory planning for compliant medical device supply
By evaluating intended purpose, classification, and regulatory scope early in development, manufacturers can create a roadmap for compliant supply.
Key Takeaway
For manufacturers planning to supply products in Australia, the most important early regulatory question is simple:
Does my product require inclusion in the Australian Register of Therapeutic Goods (ARTG)?
Confirming this requirement early helps avoid regulatory setbacks and helps ensure a smoother and compliant path to medical device supply in Australia
Click here to read more about the news.
FAQs
What is the Australian Register of Therapeutic Goods (ARTG)?
The Australian Register of Therapeutic Goods (ARTG) is the official database maintained by the Therapeutic Goods Administration that lists therapeutic goods approved for supply in Australia, including regulated medical devices.
Does My Product Need ARTG Registration in Australia?
Many manufacturers assume their product can be supplied in Australia without regulatory approval. However, if the product meets the definition of a medical device under Therapeutic Goods Administration regulations, it must generally be included in the Australian Register of Therapeutic Goods (ARTG) before supply.
When is ARTG inclusion required for medical devices?
ARTG inclusion is required before most medical devices can be legally supplied in Australia. The requirement depends on the device’s intended purpose, classification, and regulatory category under Australian medical device regulations.
How does TGA classify medical devices?
The TGA medical device classification system is risk-based and categorizes devices from Class I to Class III and Active Implantable Medical Devices. Higher-risk devices require more extensive evidence and regulatory review before inclusion in the ARTG.
What happens if a device is supplied without ARTG inclusion?
Supplying a device that requires inclusion in the Australian Register of Therapeutic Goods (ARTG) without approval may lead to regulatory enforcement actions, product removal from the market, or penalties from the Therapeutic Goods Administration.
How long does TGA medical device registration take?
The timeline varies depending on the device classification and conformity assessment pathway. The process is simpler for lower-risk medical devices compared to higher-risk medical devices.



















