









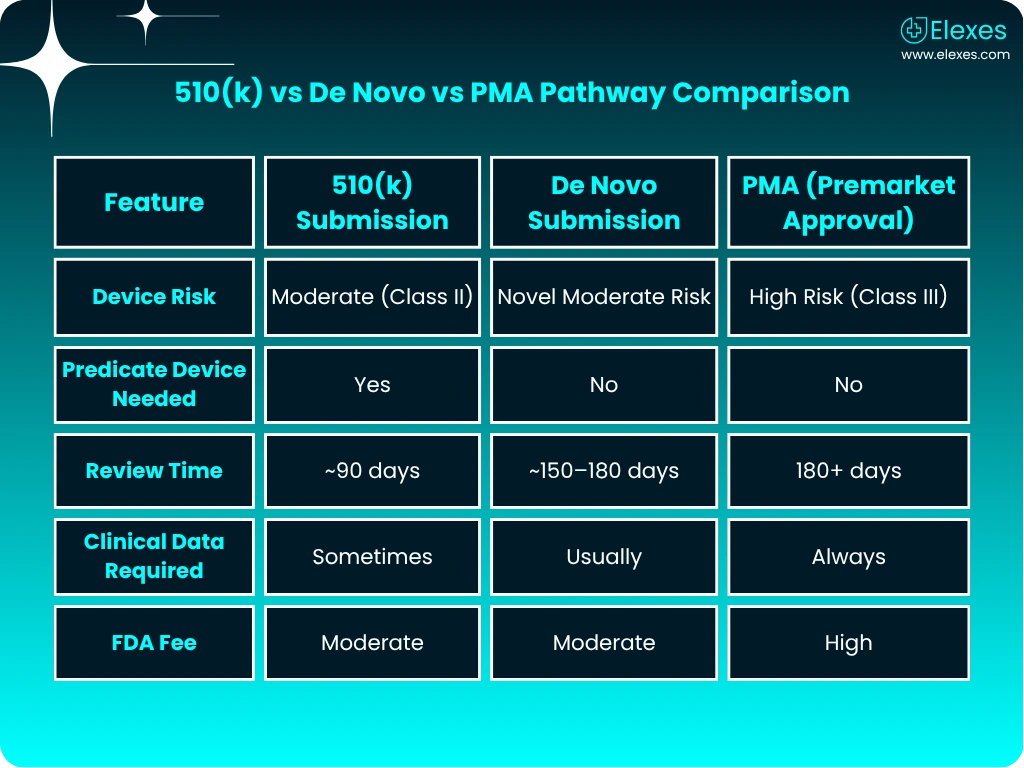
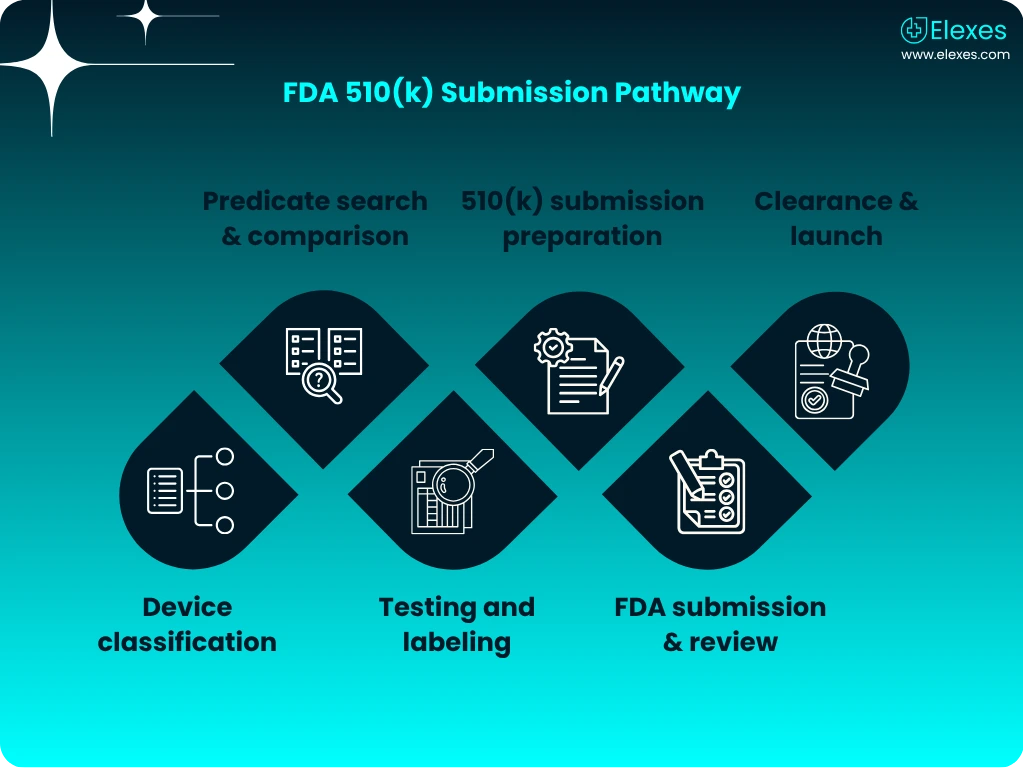





We’ve More
Than 256+ Global
Clients
The Best RA & QA Consultants

Tom Birney
CEO Masterlink, Arizona
“
Elexes gave us immense support. Their Knowledge and thoroughness were excellent. I look for all of these qualities in consulting firms
“

Diane Bryant
CEO Novasignal, Los Angeles
“Prompt and competent! Elexes should be your next regulatory consultant. Their timeline-based project plan expedited the process.
“

Daniel Kinsey
President ViDava, Florida
“Professional and easy to work with! They expedited CE Marking for our products and also created clinical evaluations, risk assessments, and provided testing/manufacturing advice.
“

Linda Pan
Sr. Exe Treedental, Hong Kong
“Having Elexes as our import-export expert was a great choice. Elexes' team answered all our questions and guided us through every step.
“

Joshua Mink
Manager Outset Medical, California
“Highly recommend Elexes! They always meet commitments and follow through on action items.
“

Michal Depa
CTO Jana Care, Massachusetts
“Highly recommended! They excelled at regulatory projects, and document control, and grasped our product's nuances quickly.
“

Philip McFerran
MD Blackrock Pharma, England
“Kudos to the Elexes team! They're always proactive and have provided invaluable support to us with import, labels, and registrations.
“

Samip Shah
VP Regulatory AliveCor, California
“Excellent communication, diligent work. There is a strong quality background and QMS norms expertise.
“

Elizabeth W
Owner Liz Inc., Arizona
“Accurate and amazing! Their regulatory-compliant fact sheets for multiple products enabled us to sell domestically and internationally without issues. Their work had an impressive turnaround time.
“

Kurt Sysock
CEO Radformation, New York
“Elexes is a go-to for all FDA regulatory and quality systems work. They outperformed every expectation!
“


























