

How to Get FDA Approval for Medical Devices: Step-by-Step Path to Clearance
In FDA regulatory terms, “approval” applies only to devices authorized through the Premarket Approval (PMA) pathway. Most medical devices legally marketed in the U.S. are FDA-cleared (510(k)) or granted De Novo classification. This guide uses “FDA approval” as a general industry phrase, while clearly distinguishing the correct regulatory terminology throughout.
If you’re trying to understand “how to get FDA approval for medical devices,” the first thing to know is that the FDA uses different premarket pathways depending on risk and whether a suitable predicate device exists. Many products receive FDA clearance (for example, through 510(k) or De Novo), while some require FDA approval through PMA. The FDA also maintains public databases of cleared and approved devices.
This guide walks you through the FDA medical device approval process end-to-end – how to determine whether you need FDA authorization, how to choose the right pathway (510(k), De Novo, or PMA), what evidence the FDA typically expects, and how timelines are calculated.
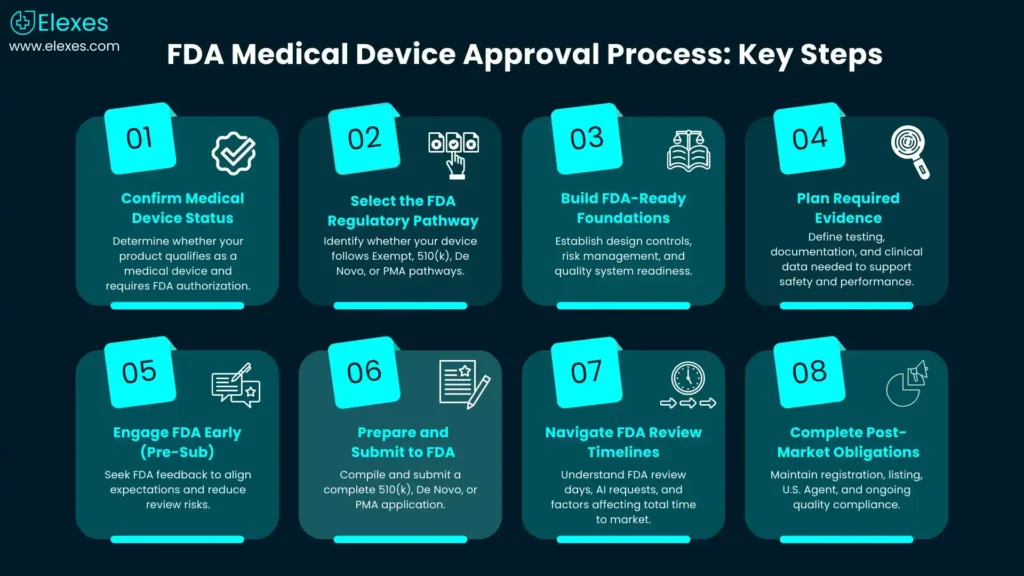
Step 1 - Confirm your product is a “medical device” and whether FDA authorization is required
Under Section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act), a medical device is defined based on intended use and includes instruments, apparatus, implements, machines, implants, in vitro reagents, or similar articles that do not achieve their primary intended purposes through chemical action within or on the body and are not dependent on being metabolized.
Do medical devices need FDA approval?
Not always. In everyday life, people say “FDA approval” when they actually mean “FDA granted the right to sell,” but the difference in regulatory language is important to know:
⦿ 510(k): FDA “clears” a device when it is found substantially equivalent to a legally marketed predicate device. Substantial equivalence requires that the device has the same intended use as the predicate and either (1) the same technological characteristics, or (2) different technological characteristics that do not raise different questions of safety and effectiveness and are supported by appropriate performance data.
⦿ De Novo: FDA grants a De Novo request to classify a novel, low-to-moderate risk device without a predicate (creating a new classification and “special controls” when applicable).
⦿ PMA: FDA “approves” Class III devices through premarket approval (PMA) based on valid scientific evidence of safety and effectiveness.
510(k) exempt devices: Some devices are exempt from 510(k) requirements (and still subject to general controls)”So the correct question is often: “Do we need a premarket submission, and if so, which one?
Practical takeaway: Your first milestone is determining whether your product requires 510(k) clearance, De Novo classification, PMA approval, or qualifies for an exemption.
Step 2 - Choose the right FDA regulatory pathway for your medical device
FDA’s “Device Approvals and Clearances” overview is a good anchor reference for how 510(k), De Novo, and PMA fit together.
510(k) clearance (Premarket Notification)
The 510(k) submission is a method to demonstrate that a device is as safe and effective as a legally marketed predicate device and does not raise different questions of safety and effectiveness.
When it fits
⦿ Predicate exists, and your intended use/technology comparison supports SE
⦿ Common for many Class II and some Class I devices (unless exempt)
De Novo classification request
De Novo is a risk-based route for new devices where no legally marketed predicate exists, and where the FDA determines that general controls alone, or general and special controls together, can provide reasonable assurance of safety and effectiveness.
When it fits
⦿ No predicate (or 510(k) is not viable)
⦿ Low-to-moderate risk novel technology
Premarket Approval (PMA)
PMA is the most rigorous pathway and is used to evaluate the safety and effectiveness of many Class III devices. PMA approval requires “valid scientific evidence,” which may include well-controlled clinical investigations, partially controlled studies, objective trials without matched controls, well-documented case histories, or significant human experience reports.
When it fits
⦿ High-risk devices (often Class III)
⦿ Clinical evidence is frequently central
Step 3 - Build your “FDA-ready” foundation before you write the submission
A successful premarket submission is usually the outcome of a compliant development process, not a collection of documents prepared in isolation. Two foundations usually lead to outcomes:
Design controls and design history (DHF)
FDA’s design control requirements (21 CFR 820.30) and associated guidance stress the importance of clearly defined design inputs and outputs, verification and validation, reviews, and design transfer controls.
What this means in practice
⦿ Clear user needs and intended use
⦿ Traceability from design inputs → outputs → verification/validation
⦿ A DHF that can be maintained and that can support changes without non-compliance
Quality system readiness (FDA compliance)
FDA has finalized the transition from the Quality System Regulation (21 CFR Part 820) to the Quality Management System Regulation (QMSR), aligned with ISO 13485:2016, with enforcement starting February 2, 2026.
Why this matters for premarket work
Even if your submission is strong, weak quality infrastructure can create downstream risk (inspection readiness, complaint handling, CAPA, design changes, and postmarket surveillance alignment).
Step 4 - Plan the evidence: what data typically supports clearance or approval?
FDA expectations are device- and risk-specific, but most submissions are built from a consistent set of evidence “modules.” Your aim is to show the device is safe and effective (PMA) or as safe/effective as the predicate (510(k)), or that general/special controls are sufficient (De Novo).
Typical evidence modules (pick what applies)
⦿ Device description & intended use (and indications for use)
⦿ Risk management aligned with hazards, mitigations, and residual risk rationale
⦿ Performance testing (bench, software, electrical safety, EMC, etc.)
⦿ Biocompatibility (if patient-contacting)
⦿ Sterilization & packaging validation (if sterile)
⦿ Software documentation (as applicable)
⦿ Labeling and instructions for use
⦿ Clinical evidence (often essential for PMA; sometimes needed for De Novo/510(k) depending on claims and risk. For 510(k) submissions, clinical data are typically required when bench or nonclinical testing alone cannot adequately demonstrate substantial equivalence)
Pro tip: The fastest way to lose time is unclear intended use/claims that force late testing changes. Align intended use, labeling, and test plans early.
Step 5 - Reduce surprises with FDA feedback (Q-Sub / Pre-Sub)
FDA’s Q-Submission Program provides mechanisms to request feedback and meetings related to medical device submissions.
When a Pre-Sub is worth it
⦿ Novel technology or unclear predicate strategy
⦿ Clinical study design questions
⦿ Nonclinical test plan alignment (especially where standards interpretation matters)
⦿ De Novo strategy and proposed special controls
A well-structured Pre-Sub can prevent misaligned test plans, avoidable AI (additional information) cycles, and rework.
Step 6 - Prepare and submit: 510(k), De Novo, or PMA mechanics
510(k) submission process (what FDA tracks and how “days” are counted)
FDA states its MDUFA goal for a 510(k) decision is 90 FDA days, excluding the time the submission is on hold during an AI request.
What to expect operationally
⦿ Administrative acceptance/validity checks
⦿ Substantive review and interactive questions
⦿ Potential AI request if key elements are missing or unclear
De Novo process and review timeline
FDA states a De Novo request is reviewed with a goal of approximately 150 FDA review days under the Medical Device User Fee Amendments (MDUFA), excluding time the submission is on hold for additional information.
PMA review process and timelines
For PMA submissions, FDA aims to complete its initial review and take regulatory action within 180 FDA days after the application is filed, excluding periods when the review clock is paused due to deficiency requests.
Submission format modernization (eSTAR and online submission)
FDA’s eSTAR program is intended to guide applicants through preparing a comprehensive submission, and FDA provides infrastructure for sending and tracking certain premarket submissions online.
Practical takeaway: Use structured templates where eligible to reduce administrative friction and avoid “incomplete” cycles.
Step 7 - Understand real timelines: what “FDA days” do (and don’t) mean FDA approval timeline for medical devices (how to interpret it)
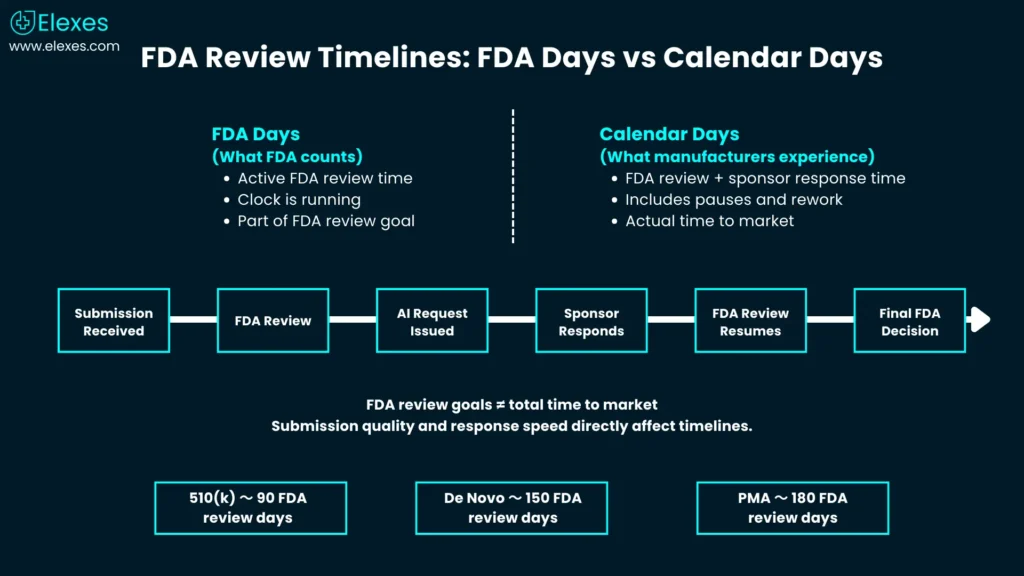
FDA’s review goals (e.g., 90 FDA days for 510(k), 150 review days for De Novo) are not the same as calendar time to market because they exclude time when FDA is waiting on you during AI holds.
What typically drives the total timeline
⦿ Predicate selection complexity (510(k))
⦿ Test execution lead times (biocomp, EMC, packaging, clinical)
⦿ Quality of initial submission (completeness and clarity)
⦿ Speed and quality of responses to AI requests
⦿ For PMA, clinical evidence and advisory committee considerations
Step 8 - After clearance/approval: don’t miss registration, listing, and ongoing compliance
Even after premarket authorization, the FDA expects certain postmarket requirements.
Establishment registration and device listing (and U.S. Agent for foreign manufacturers)
FDA requires most medical device establishments to register and list their devices annually, with registration renewal typically occurring between October 1 and December 31 each year. The FDA also notes foreign manufacturers must designate a U.S. Agent. Establishment registration and device listing do not constitute FDA approval or clearance but are administrative requirements under 21 CFR Part 807.
Maintain your quality system and inspection readiness
With QMSR enforcement beginning February 2, 2026, manufacturers should be planning to maintain an ISO 13485-aligned system in a way that also satisfies FDA expectations.
Common mistakes that delay FDA clearance or approval
“Approval” vs “clearance” confusion in strategy and messaging
The marketing terminology used by you might result in such high-risk therapeutic claims being dressed up and automatically leading you to either De Novo or PMA areas when you started with a 510(k)-appropriate profile. Follow the discipline of intended use right from the beginning.
Late predicate selection or weak substantial equivalence rationale (510(k)
Substantial equivalence does not mean only “a similar product exists”. It is a structured comparison that needs to be substantiated with evidence.
Treating quality and design controls as “later.”
Quality and Design controls are not merely a paperwork exercise; they are your means of preserving and justifying changes without losing your evidence set as well as the traceability.
Where Elexes can support you
In case your team is getting ready for U.S. commercialization and requires a lucid, defensible path, Elexes can support:
⦿ Regulatory pathway assessment (510(k) vs De Novo vs PMA vs Exemption)
⦿ Predicate strategy and submission planning
⦿ Pre-Sub (Q-Sub) preparation support
⦿ Submission readiness reviews (gap-based “missing elements” checks and completeness)
⦿ Quality system readiness aligned to FDA expectations and upcoming QMSR enforcement
⦿ Establishment registration, device listing, and U.S. Agent support (for foreign manufacturers)
The FDA Approval Process Checklist
FAQs
How long does FDA approval take?
510(k) clearance typically has an FDA review goal of 90 FDA days, De Novo requests around 150 FDA review days, and PMA timelines commonly extend beyond 6–12 months depending on clinical and review complexity.
Do I need clinical trials for 510(k)?
Not always. Most 510(k) submissions rely on bench or performance testing unless clinical performance data is essential for demonstrating equivalence.
What is the difference between PMA and 510(k)?
510(k) relies on demonstrating substantial equivalence to a predicate device, while PMA is a rigorous, data-heavy process for high-risk and novel technologies.
What’s the role of a Q-Sub or Pre-Sub?
A Q-Sub (Pre-Submission) allows sponsors to interact with the FDA before filing a submission to clarify requirements, data needs, and review expectations.
Can I market during the FDA review process?
No, you cannot market a medical device in the U.S. until the FDA issues an official clearance, grant or approval letter (510(k), De Novo, or PMA).




















