

Class III PMA Submission Guide: FDA Premarket Approval Process
The Federal Food Drug and Cosmetic (FD&C) Act established three regulatory classes for medical devices – Class I, Class II, and Class III – to ensure that these devices are safe and effective for public health. To permit only the safest and most effective medical devices into the market, the FDA has three levels of oversight based on the risk level of the devices: devices exempt from premarket notification, 510(k) Premarket Notification, and Premarket Approval (PMA). While each of these pathways requires an in-depth understanding of the regulatory framework, our focus here will be on Premarket Approval (PMA).
Elexes has guided 30+ Class III device makers through successful PMA submissions with 100% filing acceptance. For MedTech CEOs and RA Directors managing Class III launches, this guide ensures your PMA is fully aligned to FDA expectations, avoiding resubmission delays.
What is a PMA? And what kind of devices fall under the FDA PMA guidelines?
Premarket Approval (PMA) is the scientific and regulatory review by the FDA to evaluate the safety and effectiveness of Class III medical devices. These devices are the ones that support or sustain human life and present a potential of unreasonable risk of illness or injury. Approximately 10% of all medical devices enter the US market through the FDA’s PMA process and it is the second most used marketing pathway in the US. Examples of Class III devices include cardiac stents, implantable pacemakers, cochlear implants, high-frequency ventilators, etc. The FDA premarket approval is defined in CFR – Code of Federal Regulation Title 21 part 814.
Why is a Premarket Approval required?
Due to the high level of risk associated with Class III devices, general and special controls alone are not sufficient to ensure the safety of the Class III devices. Hence, as per Section 515 of the act, all Class III devices are subject to the Premarket Approval submission process.
When is a Premarket Approval required?
Unlike the other regulatory reviews, PMA is the most stringent regulatory submission. In order to determine whether a device requires a PMA or not, following considerations are important:
- An applicant needs to first identify the classification of the device. A helpful resource for such an investigation can be the Product Classification Database. The database will provide the device’s name, classification, and link to the Code of Federal Regulations if applicable
- A PMA is not required if a substantially equivalent device is available, as the submission type in such cases will be a 510(k)
- A PMA is required if the classification of the device is Class III
- A new type of device with high risk and not substantially equivalent to a medical device must require a PMA
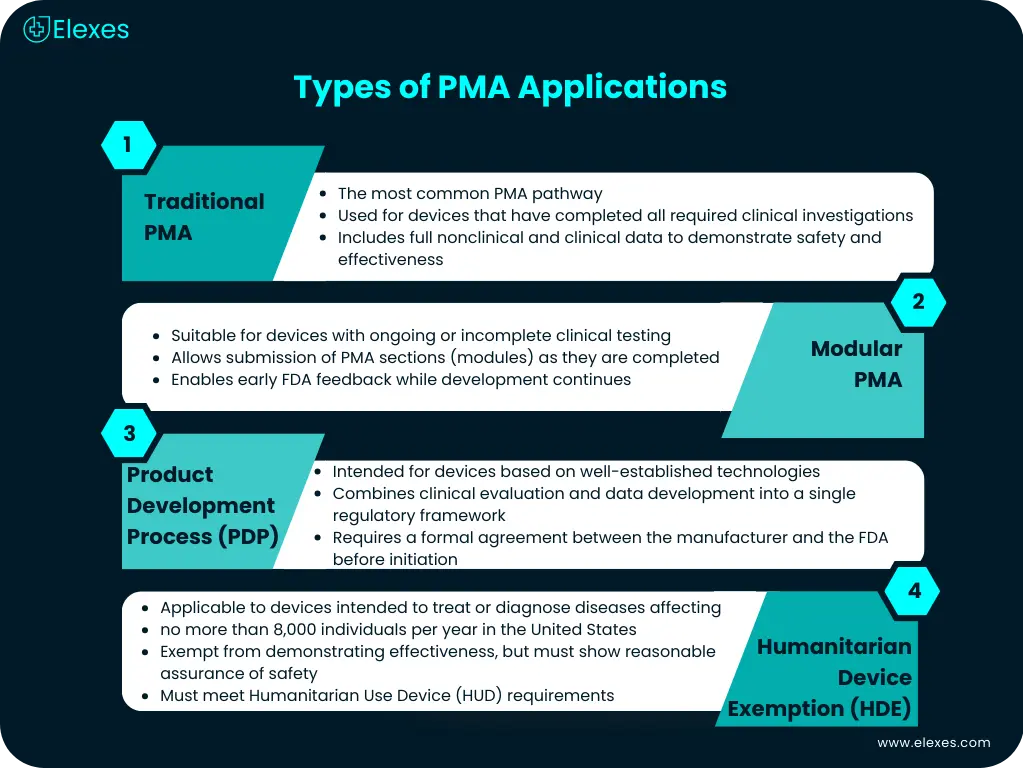
What are the types of PMA Applications?
Primarily, there are 4 types of PMA application methods:
⦿ Traditional PMA: Most common type of PMA process for the devices that have completed clinical testing.
⦿ Modular PMA: Appropriate application method for devices that have not yet completed the clinical testing.
⦿ Product Development Process Protocol: Appropriate for the devices that have well-established technology in the industry. This method merges the clinical evaluation and development of information into one regulatory mechanism and involves an agreement between the manufacturer and the FDA.
⦿ Humanitarian Device Exemption: A Humanitarian Device Exemption is the pathway for medical devices intended to serve patients in the treatment or diagnosis of a disease that affects or is manifested in not more than 8,000 individuals per year in the United States.
What are the contents and format of the PMA application?
The precise content, section and the format of a PMA application depends on the type of the PMA application, however, there is certain information which is applicable to most of the PMA application and such information is detailed below:
⦿ Cover Letter
⦿ Cover Sheet – Form FDA 3514 (Voluntary)
⦿ Table of Contents
⦿ Summary
● Indications for use
● Device description
● Alternative practices and procedures
● Marketing history
● Summary of studies
● The conclusion from the studies
⦿ Detailed description
⦿ Reference to performance standards or voluntary standards
⦿ Technical sections
● Results of nonclinical laboratory studies
● Results of clinical investigations
⦿ Justification for a PMA supported by data from a single investigator
⦿ Bibliography
⦿ One or more samples of the device or components (If required)
⦿ Proposed labeling
⦿ Environmental assessment
⦿ Financial certification or disclosure statement
● Certification: Financial Interest and Arrangements of Clinical Investigator (Form FDA 3454)
● Disclosure: Financial Interest and Arrangements of Clinical Investigators (Form FDA 3455)
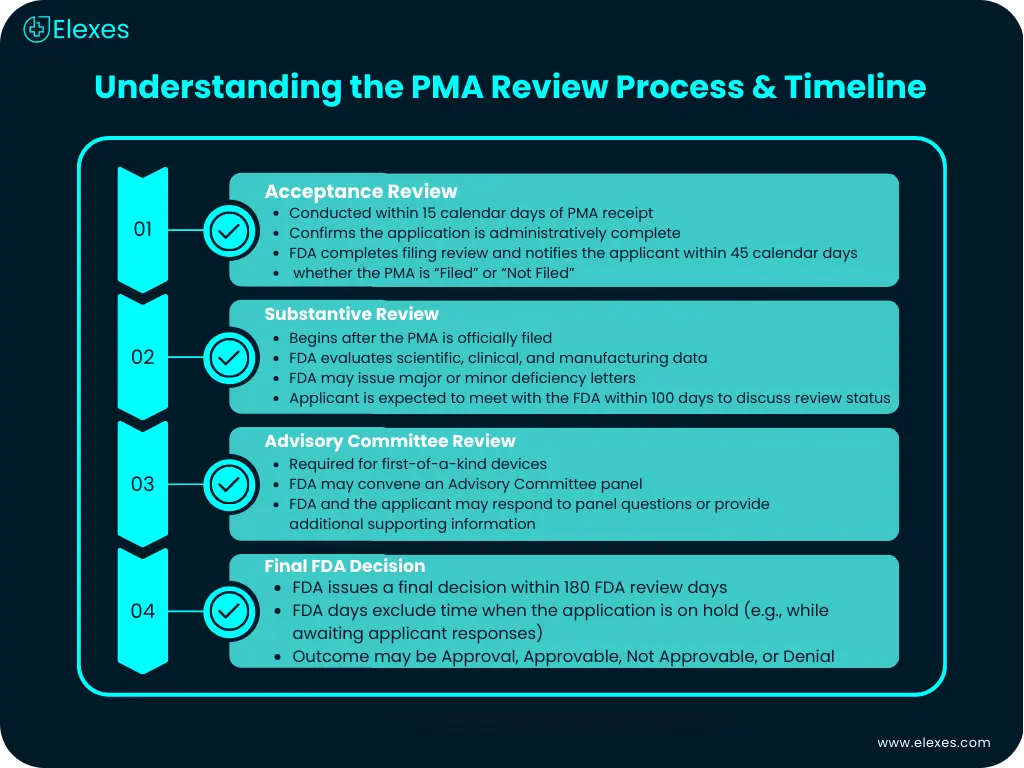
Understanding the PMA review steps and timeline
The PMA applications review is a 4-step process. The following are the primary steps involved:
Step 1: Acceptance Review
Upon receipt of the newly submitted PMA application, the FDA conducts an Acceptance Review within the 15 calendar days of the receipt. Once the application is found to be administratively complete, FDA begins filing review according to the checklist and notifies the applicant electronically within 45 calendar days of the receipt whether the PMA has been “Filed” or “Not Filed”.
Step 2: Substantive Review
FDA will conduct a substantive review of the PMA after it is accepted for filing. During the substantive review, the FDA will notify the applicant providing a major or minor deficiency letter or if any additional information is required by the FDA to complete the review. The PMA applicant is requested to meet with the FDA within 100 days to discuss the review status of the application.
Step 3: Advisory Review
The FDA might refer the PMA to an advisory committee. All PMAs for the first-of-a-kind devices are referred to this committee. During the review process, FDA may communicate with the PMA applicant or the committee to respond to questions that were raised by committee members, or to provide additional information to the panel.
Step 4: Final Decision
FDA’s final decision will be within 180 days of the application. All these days are the FDA days, and during the process when the application is put on hold etc do not get counted in here.
How much does a PMA application cost?
Annually, the FDA revisits the fees for the various applications that it reviews. As of now, following is the standard and small business fee for a PMA application.
⦿ Standard Fee: $ 374,858
⦿ Small Business Fee: $ 93,714
Is PMA really the regulatory pathway my device requires?
To assess whether or not a PMA is the right regulatory pathway for a medical device, it is important to be well acquainted with the regulatory framework in the US. Even though PMA is the most stringent, expensive, and time-consuming submission for the manufacturer, it does help in setting a high standard for market entry for potential competition.
However, since the FDA does not review a regulatory application and can reject it if it is not the right and applicable regulatory submission, any regulatory pathway if pursued without a thorough due diligence can simply result in lost time, money, and efforts. At Elexes, we have done comprehensive due diligence helping companies get an understanding of the applicable route to market entry, and on how much funds to raise and what efforts to put in to bring their products to the market.
How do I ensure that my PMA is a success?
Success of a PMA largely depends on the quality of data and evidence submitted that support the safety and efficacy of the subject device, and the manner in which information is presented. Given that a PMA submission is a highly complex and lengthy submission, it is important that all stakeholders whose departments provide information for this are well aligned on what is going into the application and what is the company seeking an approval for.
This requires continuous checks for consistency, productive cross functional interactions and ensuring all data is being effectively leveraged and showcased. Having done several such submissions, Elexes Team can come handy during such cumbersome efforts and can make the entire submission data gathering and compilation efforts highly efficient.
Helping medical device companies bring their products to the market and continually assisting with post market compliance is our passion.
If you are facing any problems in regulatory, quality, or clinical, or want additional support or looking for staff augmentation
FAQs
What is a PMA submission for medical devices?
A PMA (Premarket Approval) submission is the FDA’s rigorous regulatory pathway for approving Class III medical devices based on scientific evidence of safety and effectiveness.
When is a PMA required instead of a 510(k)?
A PMA is required when a medical device is classified as Class III PMA and there is no medical device available and the indications for use are specific.
What are the main components of a PMA application?
A PMA typically includes nonclinical and clinical data, manufacturing details, device labeling including Instructions for Use (IFU) and risk analysis documentation.
How long does the FDA take to review a PMA submission?
The FDA PMA review timeline can take 180 days or longer, with key milestones such as Day-45 filing decision, Day-100 meeting, and, for some devices, an advisory panel review.
How can Elexes help with a PMA submission?
Elexes provides end-to-end support including submission document preparation, clinical data review, QMS readiness, and communication with the FDA to streamline PMA approvals.



















