
Introduction
Uncontrolled changes are one of the fastest routes to FDA enforcement action, device recalls, and failed audits. For medical device manufacturers, QA/RA teams, startups, and SaMD and IVD developers, a single undocumented modification can compromise device safety, destabilize a quality management system (QMS), and trigger regulatory consequences that take years to resolve.
Medical device change control is the formal, documented process that prevents this — ensuring every modification to a device, process, document, or system is reviewed, assessed, and approved before implementation.
The FDA Warning Letter issued to iRhythm Technologies in May 2023 shows exactly what this looks like in practice: hardware, firmware, and algorithm changes implemented without a new 510(k) submission and without adequate validation. Poor change management has similarly driven documented recalls and audit non-conformances across global markets.
This article breaks down change control under ISO 13485 — what it covers, how the process works, and where teams most often go wrong.
Key Takeaways
- Medical device change control is a structured process to evaluate, approve, and document modifications before implementation, as required by ISO 13485 Section 4.1.4.
- It covers document, design, production/process, and regulatory changes — each requiring proportionate review.
- The core process: initiation → impact assessment → risk review → regulatory assessment → implementation → documentation and monitoring.
- Poorly managed change control is a leading cause of audit non-conformances and regulatory rejections — proper execution protects both product integrity and your QMS.
- Retrospective change approval after implementation violates both FDA and ISO 13485 requirements; compliance cannot be applied after the fact.
What Is Medical Device Change Control?
Change control is the systematic process of identifying, evaluating, approving, and recording modifications to any element of a medical device's design, production, documentation, or quality system. The goal: ensure changes do not negatively impact safety, performance, or regulatory approval status.
Change Control vs. Change Management
These two terms are often used interchangeably, but they're not the same:
- Change management — the broader organizational discipline for handling transitions, communication, and adoption
- Change control — the specific, documented, gated process within the QMS that formally evaluates and authorizes each change before it takes effect
Why It Matters
Change control is not bureaucratic red tape imposed solely for compliance. In practice, seemingly minor modifications can have far-reaching consequences.
Consider a real example: a company changed what it believed to be a non-functional feature of a device component. That modification disrupted a customer's assembly line because the "non-functional" feature was actually a critical alignment reference. Formal change control protects companies from these cascading risks — not just regulators.
Types of Changes That Require Formal Change Control
Change control applies to four broad categories, and the depth of the process should be proportional to the risk and scope of each change. An administrative document correction requires far less rigor than a design change to an implantable device — here's how each category works.
Document Changes
Any revision to a controlled document — SOPs, work instructions, forms, quality procedures — must go through change control. The same functions that approved the original document must review and approve the revision.
Document changes follow a more streamlined process than design changes. Purely administrative edits (such as clarity corrections) carry less rigor than process-driven updates that affect how work is performed.
Design Changes
Changes to the device itself — specifications, materials, labeling, packaging, or supplier components — require full change control. These modifications can directly affect device safety, and regulators treat them accordingly. A practical test: Does the change impact form, fit, or function? If yes, it is a design change.
Design changes originally included in a regulatory submission (510(k), CE marking technical file) may require regulatory notification or a new submission before implementation. The FDA's guidance on when to submit a 510(k) for a device change provides a formal decision framework for this step.
Production and Process Changes
Modifications to manufacturing processes, facilities, inspection criteria, equipment, cleanroom classification, or validated systems require change control. Process changes often trigger revalidation under ISO 13485 Section 7.5.6 and must be approved per document control requirements before implementation.
Regulatory and Supplier Changes
Two frequently overlooked change types deserve attention:
- Entering a new market or changing an authorized representative/sponsor constitutes a regulatory change — one that must be reviewed and documented through formal change control.
- Supplier-initiated changes fall under ISO 13485 Section 7.4.2, which requires written agreements ensuring suppliers notify the organization of any changes to purchased products before implementation. Quality agreements typically formalize this through explicit change notification clauses.

How the ISO 13485 Change Control Process Works: Step by Step
The change control process follows a gated, multi-step structure regardless of organization size. Each phase must be completed and documented before the next begins.
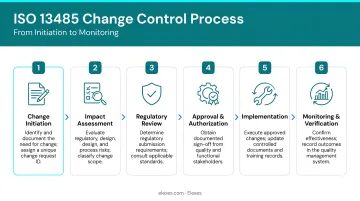
Step 1: Change Initiation
Any person in the organization can initiate a change by submitting a formal change request (also called a change order). The request should document:
- Origin of the change
- Reason it is needed
- Submitter's identity
- Scope or cost implications (if known)
The request must be specific enough for the change control board to make a preliminary decision.
Step 2: Impact Assessment
Once initiated, the change is evaluated for its impact across all affected areas:
- Other device components
- Current inventory and devices already in the field
- Marketing materials and labels
- Validation status
- Supplier and customer notification obligations
This is a cross-functional exercise involving quality, engineering, supply chain, regulatory, and commercial teams.
Step 3: Risk Assessment
Every change must be evaluated for its effect on device risk. This may involve:
- Updating the product FMEA
- Opening a new risk analysis
- Revising the risk management file
A preliminary risk assessment may be conducted early to decide whether to proceed. A final risk assessment is then performed before implementation to confirm no new risks have been introduced.
Step 4: Regulatory Review
Skipping or underestimating regulatory review is one of the most common sources of compliance failures. The regulatory impact must be assessed before implementation:
- Does the change require a new 510(k), PMA supplement, or EU MDR notified body notification?
- Does it trigger requirements from other regulatory authorities?
- For post-market devices, are customer notifications required?
The FDA's guidance document on deciding when to submit a 510(k) for a change provides a formal decision framework. For organizations managing multi-market submissions, regulatory consultants with cross-jurisdictional experience — such as Elexes — can accelerate this assessment significantly.
Step 5: Implementation and Verification/Validation
Once all assessments are complete and approvals obtained, the team executes the change. This may include:
- Updating work instructions, BOMs, and device master records
- Performing re-validation
- Training affected personnel
- Notifying suppliers or customers
A clear effective date must be established and communicated. Verification and validation activities must be documented as part of the change record.
Step 6: Documentation and Post-Implementation Monitoring
Every decision, approval, justification, and assessment must be documented to form a complete, traceable change record that any external auditor can follow at any point in the device lifecycle.
Post-implementation, the organization should:
- Monitor whether the change achieved its intended outcome
- Reassess risk if needed
- Address any emerging nonconformances
- Capture lessons learned to improve future change control activities
Key Factors That Affect Change Control Effectiveness
Change Scope and Classification
The scope and classification of a change determine how demanding the review process needs to be. A risk-proportionate approach means minor document revisions and major design changes should not follow the same level of rigor. Organizations should define clear criteria for change classification — for example, major vs. minor — directly in their change control SOP.
Documentation Quality and Traceability
Incomplete change records are one of the most frequently cited ISO 13485 non-conformances during certification and surveillance audits. Every change record must include:
- Description of the change
- Affected documents
- Approver signatures and approval dates
- Effective date
Missing justifications, unsigned approvals, or absent regulatory assessment evidence frequently trigger audit findings.
Cross-Functional Participation
Strong documentation addresses what was changed — but cross-functional participation determines whether the full impact was actually understood. Change control boards that exclude regulatory, supply chain, or commercial stakeholders during impact assessment routinely miss downstream consequences.
For organizations pursuing multi-market approval, each jurisdiction sets its own threshold for when a change triggers a notification or new submission:
- FDA: May require a new 510(k) or PMA supplement depending on change significance
- EU MDR: Changes affecting safety or performance can require Notified Body review
- Health Canada: Device changes may necessitate an amendment to the existing license
- MDSAP: Impacts all five participating regulatory authorities simultaneously

Common Misconceptions and Mistakes in Change Control
Three misunderstandings consistently create audit findings, enforcement risk, and QMS gaps. Here's where teams most often go wrong.
Change Control Only Applies to "Big" Changes
Document updates, process modifications, supplier substitutions, and regulatory status changes all require formal control under ISO 13485 — not just hardware or design changes. Teams that reserve change control for major modifications routinely accumulate minor uncontrolled edits that collectively compromise QMS integrity.
Changes Can Be Documented After the Fact
Changes must be evaluated and approved before implementation, not retrospectively documented afterward. This is one of the most common audit findings and can indicate systemic QMS failure to a notified body or FDA inspector.
Real-world consequences are well-documented. Both the iRhythm Technologies warning letter and the Rolence Ent. warning letter cited failures to validate or approve changes before use, resulting in enforcement action.
"Approved" Means "Complete"
Approval by the change control board is authorization to proceed — not confirmation the change will be fully implemented. Many changes are later abandoned due to validation failures, unforeseen costs, or regulatory barriers uncovered during execution.
Proper change control procedures account for this by requiring a final close-out review before marking any change as effective.
Frequently Asked Questions
What is change control in a medical device?
Change control in medical devices is the formal process of reviewing, assessing, and approving any modification to a device's design, documentation, process, or quality system before implementation. This ensures changes do not compromise device safety, performance, or regulatory approval status.
What are the steps of the change control process?
The core steps are change initiation, impact assessment, risk assessment, regulatory review, implementation with verification or validation, and documentation with post-implementation monitoring. The depth of each step should reflect the risk and scope of the change.
What are the criteria for change control?
Any modification to a controlled document, device design (affecting form, fit, or function), production process, facility, validated system, supplier, or regulatory status triggers change control. The criteria should be defined in the organization's change control SOP to ensure consistent classification.
What is the 7.3 clause of ISO 13485?
ISO 13485 Clause 7.3 covers Design and Development, including Section 7.3.9 (Control of Design and Development Changes), which requires that any changes to device design be identified, documented, reviewed, verified or validated, and approved before implementation.
What does clause 7.4.2 of ISO 13485 provide details of?
ISO 13485 Clause 7.4.2 covers Purchasing Information and requires organizations to notify suppliers of any changes to purchased products or services before implementation. This is typically addressed through a quality agreement with a formal change notification clause.
What is the ISO 13485 document control clause?
ISO 13485 Clause 4.2.4 governs document control and requires that changes to documents be identified, reviewed, and approved by the same functions that performed the original review. Change records must be retained to ensure traceability of every document revision throughout the QMS.


