
Medical device approval is not the finish line—it marks the beginning of ongoing regulatory obligations. Once a device reaches real-world patients, manufacturers face mandatory, continuous post-market surveillance (PMS) requirements to ensure safety and effectiveness across diverse populations and use conditions. This oversight extends far beyond the controlled environment of premarket clinical trials.
Many manufacturers treat PMS as a checkbox exercise or underestimate its complexity across multiple regulatory jurisdictions—FDA, EU MDR, ISO standards, Health Canada, MHRA, TGA, and others. This approach leads to costly non-compliance, including product seizures, market withdrawals, and suspension of regulatory certifications.
This guide covers the essential elements of postmarket surveillance: the regulatory definition and purpose of PMS, global regulatory requirements across major markets, the mandatory components of a compliant PMS plan, the end-to-end PMS process workflow, the critical distinction between PMS and PMCF, and the consequences of non-compliance.
Key Takeaways
- PMS is the active, ongoing monitoring of a device's safety and performance after market entry
- FDA requires PMS for higher-risk Class II and Class III devices under 21 CFR Part 822; EU MDR mandates PMS for all CE-marked devices
- A compliant PMS plan covers surveillance objectives, data collection methods, statistical analysis, and reporting timelines
- PMCF is a clinical subset of PMS required under EU MDR, not synonymous with FDA's 522 surveillance orders
- Gaps in compliance can trigger misbranding findings under FDA law or CE mark suspension under EU MDR
What Is Postmarket Surveillance and Why Does It Matter?
Postmarket surveillance is the active, systematic, scientifically valid collection, analysis, and interpretation of data about a marketed medical device, as defined in 21 CFR §822.3. The term "active" is critical—PMS extends beyond passively waiting for complaints or adverse event reports.
Why Premarket Data Is Insufficient
Premarket clinical trials involve relatively small, carefully selected patient populations. These controlled studies often exclude patients with comorbidities, pediatric or geriatric populations, and edge-use cases that reflect real-world conditions. FDA's 522 program exists precisely because premarket data alone cannot capture all real-world risks and performance variations.
PMS fills that gap — collecting data across diverse real-world populations, extended use periods, and clinical settings that controlled trials cannot replicate.
Passive vs. Active Surveillance
Regulators distinguish between two primary surveillance modes:
- Passive surveillance — voluntary adverse event reporting systems like FDA's MedWatch or spontaneous reporting databases
- Active surveillance — manufacturer-initiated studies, registries, patient follow-up programs, and proactive data collection efforts like FDA's Sentinel System
Regulators globally are pushing manufacturers toward more active, systematic surveillance methods rather than relying solely on passive reporting.
PMS as a Living System
That active orientation makes PMS a continuous quality loop, not a one-time study. Under EU MDR Article 83, manufacturers must feed PMS findings back into core quality processes:
- Update benefit-risk determinations as real-world evidence accumulates
- Strengthen risk management files when new hazards emerge
- Revise clinical evaluations to reflect post-launch performance data
- Initiate corrective and preventive actions (CAPA) when safety signals appear
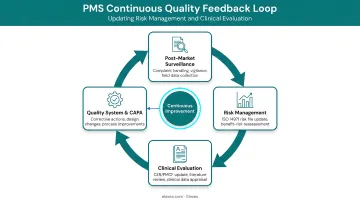
ISO 14971:2019 Clause 10 reinforces this obligation, requiring manufacturers to collect and review production and post-production information for safety relevance. In practice, PMS data drives labeling updates, design improvements, and risk file revisions at every stage of a device's commercial life.
Postmarket Surveillance Regulatory Requirements: FDA, EU MDR, and More
FDA Requirements: Section 522 and 21 CFR Part 822
The FDA can require postmarket surveillance for Class II and Class III devices meeting specific statutory triggers under Section 522 of the FD&C Act:
Four mandatory triggers:
- Device failure likely to have serious adverse health consequences
- Implanted for more than one year
- Life-sustaining or life-supporting device used outside a user facility
- Device with significant pediatric use
Critical timelines:
- Manufacturers must submit a PMS plan within 30 days of receiving a 522 order
- Surveillance must commence within 15 months of the order
- Failure to comply renders the device misbranded under 21 CFR §502(t)(3)
EU MDR: Broader Scope and Mandatory for All Devices
Under EU MDR Regulation 2017/745, PMS is mandatory for all medical device classes—not just high-risk devices. The regulation requires a proactive, systematic process integrated into the Quality Management System.
Key EU MDR PMS documentation:
| Document Type | Device Class | Update Frequency | Legal Basis |
|---|---|---|---|
| PMS Plan | All classes | Continuous updates | Article 84 |
| PMS Report | Class I | When necessary | Article 85 |
| PSUR (Periodic Safety Update Report) | IIa, IIb, III | IIa: every 2 years; IIb/III: annually | Article 86 |
The MDCG 2025-10 guidance provides the current best practice framework for PMS implementation under EU MDR.
ISO Standards: Quality System Integration
ISO 13485:2016 Clause 8.2.1 requires manufacturers to maintain a documented procedure for gathering and reviewing post-production information. This data must feed into corrective action and risk management processes.
ISO 14971:2019 Clause 10 requires manufacturers to collect production and post-production information and review it for safety relevance. Any findings must feed back into the risk management file, updating controls as needed. Together, these two standards form the quality and risk management backbone of a compliant PMS system.
Where ISO 13485 defines the procedural infrastructure, ISO 14971 ensures PMS data actively updates device risk profiles—connecting surveillance activity to real safety decisions.
Global PMS Requirements
Beyond FDA and EU MDR, manufacturers selling across multiple markets face jurisdiction-specific vigilance obligations:
- Health Canada — Medical Devices Regulations SOR/98-282 establishes incident reporting timelines and foreign risk notification requirements
- MHRA (UK) — The Yellow Card Scheme handles adverse incident reporting, with dedicated vigilance reporting requirements for device manufacturers
- TGA (Australia) — The Incident Reporting and Investigation Scheme (IRIS) manages adverse event reporting and corrective actions
- SFDA (Saudi Arabia) — The National Center for Medical Devices Reporting (NCMDR) coordinates post-market safety surveillance and follow-up

Elexes has managed PMS compliance programs across these jurisdictions for 100+ global clients, helping manufacturers build unified surveillance systems that satisfy multiple regulatory bodies simultaneously.
What Must a Postmarket Surveillance Plan Include?
FDA PMS Plan Requirements (21 CFR §822.10)
A compliant FDA PMS plan must include:
- Surveillance objectives tied to the specific question being addressed
- Study subjects and variables/endpoints
- Surveillance methodology (prospective, retrospective, observational, etc.)
- Sample size and units of observation
- Data sources (hospital records, registries, EHRs)
- Data collection plan and forms
- Monitoring procedures
- Estimated surveillance duration
- Statistical analysis plan
- Interim and final report schedule
The FDA reviews submissions within 60 days and may issue approval orders, approvable letters with conditions, or disapproval notices requiring resubmission.
EU MDR PMS Plan Requirements (Article 84)
Under EU MDR Annex III, the PMS plan must address proactive data collection across multiple sources:
- Serious and non-serious incidents
- Field safety corrective actions (FSCA)
- Trend reports and analysis thresholds
- Published literature reviews
- Implant registries and clinical databases
- Customer complaints and feedback
- Publicly available information about similar devices
- PMCF plan or documented justification for its absence
The plan must reference the device's intended purpose and be updated continuously, not just at fixed intervals.
Both frameworks also expect manufacturers to draw on sources beyond formal complaint channels. Sales and distribution data, social media monitoring, and real-world use reports are increasingly scrutinized during audits and reviews — yet they're often missing from first-draft PMS plans.
Designated Person and Investigator Requirements
Under 21 CFR §822.3(b) and §822.9, the PMS plan must name a designated person with appropriate qualifications to conduct or supervise surveillance. FDA reviews these qualifications as part of plan approval.
Under EU MDR, PMS is integrated into the Quality Management System under a Person Responsible for Regulatory Compliance (PRRC), rather than designated to a single investigator.
In both cases, the qualifications and accountability structure of whoever owns PMS are evaluated during regulatory review — making this one of the more consequential decisions in plan preparation.
The PMS Process: From Data Collection to Regulatory Reporting
Step-by-Step PMS Workflow
- Receive PMS order or trigger obligation — FDA issues 522 order, or manufacturer initiates per EU MDR
- Define surveillance question — identify specific safety or performance concerns
- Design and submit PMS plan — within 30 days for FDA orders
- Regulatory review and approval — FDA reviews within 60 days; EU MDR plans integrate into QMS
- Initiate surveillance activities — within 15 months for FDA orders
- Collect and analyze data — systematically gather data from all planned sources
- Submit interim reports — per approved plan schedule
- Submit final report — after surveillance period concludes
- Feed findings into risk management and CAPA — update risk files, labeling, and design controls
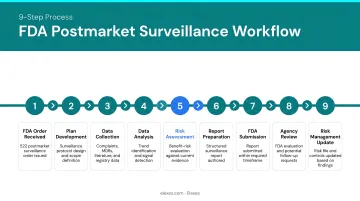
Under FDA rules (21 CFR §822.15), prospective surveillance periods can be ordered for up to 36 months; longer periods require mutual agreement.
Reporting Obligations
FDA (21 CFR Part 822 & 803): Manufacturers must submit interim and final reports per the approved plan schedule under 21 CFR §822.38. Unexpected serious adverse events require separate reporting via Medical Device Reporting (MDR, 21 CFR Part 803).
EU MDR (Article 87): Serious incidents must be reported through the Article 92 electronic system under Article 87. Required timelines vary by severity:
- 15 days — serious incidents
- 10 days — death or unanticipated serious deterioration in health
- 2 days — serious public health threats
Note that PSUR periodic reporting is separate from reactive incident reporting and follows the Article 86 schedule — the two obligations run in parallel, not in sequence.
Records Retention and Inspection Readiness
Under 21 CFR §822.33, manufacturers must retain all PMS records—including correspondence, data, deviations from the plan, and consent documents—for 2 years after FDA acceptance of the final report.
FDA can inspect PMS records during routine inspections, recall investigations, or PMS-specific inspections under 21 CFR §822.35. Manufacturers should ensure records are organized and retrievable within 72 hours of an inspection request.
PMS vs. PMCF: Key Differences Explained
Defining the Distinction
PMS (Postmarket Surveillance) is the broad, overarching system for monitoring device safety and performance using all available data sources—complaints, literature, registries, adverse event databases, sales records, and more.
PMCF (Post-Market Clinical Follow-up) is a specific clinical activity within PMS that collects new clinical data on a device through studies, surveys, or registries. MDCG 2020-7 defines PMCF as a continuous process to proactively collect and evaluate clinical data from use of a CE-marked device to confirm safety and performance and update clinical evaluation.
Key point: PMS without PMCF can meet FDA requirements. EU MDR mandates PMCF (or documented justification for its absence) for all device classes.
When Is PMCF Required Under EU MDR?
PMCF is specifically required when:
- Novel technology or clinical procedure is involved
- High-risk device classes (IIb, III) or implantable devices
- Insufficient clinical data from premarket studies
- Devices used in high-risk populations (pediatric, geriatric)
- Residual uncertainty about long-term safety or performance

The PMCF Plan and PMCF Evaluation Report are separate documents within the Technical Documentation.
PMS vs. Real World Evidence (RWE)
Real World Evidence (RWE) refers to clinical evidence derived from real-world data sources like electronic health records, claims databases, and patient registries. RWE is not a regulatory system. It is a data source that feeds into both PMS and PMCF activities.
FDA's 522 Postmarket Surveillance Studies Guidance recognizes fulfilling 522 orders using relevant and reliable real-world data sources. Regulators now accept RWE as part of PMS submissions when the data is validated and relevant to the device's intended use.
Consequences of PMS Non-Compliance and Common Pitfalls
FDA Regulatory Consequences
Failure to submit a PMS plan, having a plan disapproved without resubmission, or failing to conduct surveillance per the approved plan all constitute violations of Section 522 of the FD&C Act.
Legal consequences under 21 CFR §822.20:
- Device is considered misbranded under §502(t)(3)
- Product seizure
- Injunctions
- Civil monetary penalties
- Criminal prosecution
EU MDR Consequences
Failure to maintain a compliant PMS system can result in:
- Suspension or withdrawal of CE certificate by the Notified Body
- Removal from the EUDAMED database
- Recall orders from national competent authorities
- Market withdrawal across EU member states
Enforcement risk is rising as EUDAMED's four first modules become mandatory on 28 May 2026, increasing data visibility across EU member states and leaving gaps in PMS documentation harder to obscure.
Common Pitfalls to Avoid
These are the most common compliance gaps manufacturers encounter:
- Static documentation mindset — treating PMS as a one-time document rather than a living system
- Failure to update after changes — not revising the PMS plan after design changes or new safety signals
- Inadequate data sources — relying solely on passive complaint data without systematic literature surveillance
- Multi-market documentation gaps — underestimating documentation requirements for compliance across multiple jurisdictions
Best practice: Conduct periodic internal PMS audits and treat each audit cycle as an opportunity to update data sources, not just verify documentation. Engaging a regulatory consultant before an inspection — rather than after a finding — gives manufacturers time to close gaps on their own terms.
Elexes has supported 250+ global projects with a 90% audit clearance rate, helping manufacturers build PMS systems that hold up under regulatory scrutiny.
Frequently Asked Questions
Does the FDA require post market surveillance?
Yes. The FDA can require PMS for Class II and Class III devices meeting specific criteria under Section 522 of the FD&C Act and 21 CFR Part 822. Devices that fail with serious adverse health consequences, are implanted for more than one year, are life-sustaining devices used outside facilities, or have significant pediatric use may be subject to 522 orders. Class I devices are generally exempt.
What is a post market surveillance plan?
A PMS plan is a formal document submitted to the FDA within 30 days of a PMS order, or maintained in Technical Documentation under EU MDR. It defines the surveillance question, methodology, data sources, analysis approach, and reporting schedule — and must comply with 21 CFR §822.10 (FDA) or EU MDR Annex III.
How long does post market surveillance last?
Under FDA rules, the agency may order prospective surveillance for up to 36 months; longer durations require manufacturer agreement. Under EU MDR, PMS is continuous throughout the device's lifetime on the market and does not have a defined end date—it remains an ongoing obligation as long as the device is CE-marked.
What is the difference between PMCF and PMS?
PMS is the broad surveillance system using all available data sources to monitor device safety and performance. PMCF is a specific clinical sub-activity within PMS that collects new clinical evidence through studies or registries. PMCF is an EU MDR-specific requirement and is not directly equivalent to FDA's 522 studies.
What ISO standard covers post market surveillance?
ISO 13485:2016 Clause 8.2.1 requires a documented post-production information procedure. ISO 14971:2019 requires PMS data to feed back into the risk management process. Both standards apply across FDA, EU MDR, and most global markets — meaning compliance with one supports compliance with the others.
How much does post market surveillance cost?
Costs vary based on device class, target markets, study complexity, and whether PMCF is required. Passive surveillance programs may require only internal resource allocation, while active programs involving registries or prospective studies can reach hundreds of thousands of dollars. Efficient program design — often with outside regulatory support — can reduce costs substantially for multi-market programs.


