
Introduction
EU MDR 2017/745 represents a fundamental shift in how manufacturers approach post-market surveillance. Unlike its predecessor, the Medical Device Directive (MDD), the EU MDR mandates a proactive, continuous PMS system as a core component of every manufacturer's Quality Management System — not a one-time compliance checkbox.
This requirement is enshrined in Article 10(10), which lists PMS as a mandatory QMS element. That distinction matters: PMS is no longer a documentation exercise completed at approval. It is an ongoing obligation tied directly to market access.
The consequences of non-compliance are severe and immediate. Manufacturers who fall short face:
- Regulatory action from Notified Bodies or Competent Authorities
- Forced market withdrawal of CE-marked devices
- Delayed or denied CE certification renewals
- Lasting reputational damage in the EU market
With over 21,714 MDD/AIMDD legacy certificates in existence as of 2022 and only 14% transitioned to MDR, most manufacturers still face a full PMS system overhaul.
This guide covers exactly what that overhaul requires: the legal basis for PMS under EU MDR, required system components, PMS plan structure, reporting timelines by device class, QMS integration, and the compliance gaps most manufacturers need to close.
Key Takeaways
- EU MDR Articles 83–86 require all manufacturers to maintain a live, proactive PMS system throughout the entire device lifetime
- Required outputs include a written PMS plan, defined data collection methods, and a PMS Report (Class I) or PSUR (Class IIa, IIb, III)
- PMS data feeds directly into risk management, clinical evaluation, labelling, and CAPA — it cannot operate in isolation
- Post-Market Clinical Follow-up (PMCF) applies to higher-risk devices unless you can document a clear, justified exemption
- Non-compliance triggers Notified Body scrutiny, CE mark suspension, and Competent Authority enforcement — up to market withdrawal
Why EU MDR Post-Market Surveillance Compliance Matters
The Legal Mandate
Article 10(10) of EU MDR 2017/745 requires all manufacturers to implement and maintain a PMS system as an integral part of their QMS. This is not optional best practice — it's a legal obligation applying to all device classes from Class I through Class III. Article 83(1) reinforces this by stating that manufacturers must "plan, establish, document, implement, maintain and update a post-market surveillance system in a manner that is proportionate to the risk class."
The Safety Rationale
PMS exists to continuously verify that a device performs safely and effectively in real-world conditions, beyond what pre-market clinical evaluations can capture. Real-world use surfaces risks — new side effects, misuse patterns, performance drift across diverse patient populations and operator skill levels — that controlled trials routinely miss.
Reactive vs. Proactive PMS
A critical distinction separates compliant manufacturers from those at audit risk: reactive versus proactive surveillance. Reactive manufacturers wait for complaints to arrive. Proactive manufacturers actively seek feedback from literature, registries, and users. MDCG 2025-10 guidance explicitly requires a "proactive and systematic" approach under Annex III. Reactive-only approaches are a common cause of audit findings.
Commercial and Market Access Implications
For Class IIb and III devices, PSURs are submitted directly to Notified Bodies and increasingly via EUDAMED. This means PMS performance is now visible to regulators and can directly affect certification renewal outcomes. Poor PMS documentation or delayed PSUR submissions can result in conditional certifications or renewal delays, directly impacting market access.
How EU MDR Raised the Bar from MDD
Those commercial stakes stem directly from how EU MDR tightened the rules. While MDD had PMS requirements, EU MDR went further in four concrete ways:
- Codified mandatory structure for PMS plans, reports, and evaluation documents
- Increased reporting frequency — annual PSURs for Class IIb/III, periodic for lower classes
- EUDAMED transparency — submissions now visible to regulators across member states
- Mandatory PMCF for higher-risk devices, requiring ongoing clinical data collection

Regulation (EU) 2023/607 extended transition deadlines to 31 December 2027 (Class III/IIb implantable) and 31 December 2028 (Class IIb/IIa/Class I sterile or measuring). PMS requirements apply immediately, however — even to legacy devices still operating under MDD certificates during the transition period.
According to TEAM-NB data, more than 75% of legacy certificates had no MDR application filed as of end 2021 — meaning the majority of manufacturers were still building PMS systems from scratch while the compliance clock was already running.
Key Components of the EU MDR PMS System
System Architecture
Under Article 83, the PMS system is not a single document but a collection of interconnected processes governed by the manufacturer's QMS:
- Data collection
- Data analysis
- Conclusion-drawing
- Corrective action
- Output reporting
These processes must function continuously throughout the device lifecycle.
The PMS Plan (Article 84 / Annex III)
The PMS plan is the foundational document that defines:
- What data to collect
- From which sources
- At what frequency
- Using which methods
This must be part of the technical documentation for all classes (with specific provisions for custom-made devices) and must be established during product development, not after market launch. Notified Bodies review the PMS plan during technical file assessment. Inadequate plans are a leading cause of certification delays — a risk easily avoided with early-stage planning.
The PMS Report and PSUR (Articles 85–86)
Class I devices require a PMS Report available to Competent Authorities on request, updated as needed.
Class IIa, IIb, and Class III devices require a Periodic Safety Update Report (PSUR) submitted to the Notified Body. PSURs must include:
- Benefit-risk conclusions
- PMCF findings
- Sales and patient exposure data
- Trend analysis
- Updated risk assessments
PSURs carry significantly more content and scrutiny than PMS Reports — and PMCF findings are a core input to that structure.
Post-Market Clinical Follow-up (PMCF)
PMCF is the clinical arm of PMS, governed by Annex XIV Part B of EU MDR. It requires a documented PMCF plan specifying how the manufacturer will proactively gather clinical data post-launch through:
- Patient registries
- User surveys
- Clinical studies
- Literature reviews
If a manufacturer determines PMCF is not required, a written justification must be included in the PMS plan.
Vague or insufficient justifications are among the most common Notified Body findings during technical file review.
Vigilance and Trend Reporting
Vigilance under Articles 87–88 is event-driven and reactive — but it feeds directly into the broader PMS system. Beyond serious incidents, PMS must also track:
- Non-serious incidents
- Expected undesirable side effects
Article 88 requires reporting to Competent Authorities when a statistically significant increase in incident frequency or severity is detected. This trend reporting approach must be defined in the PMS plan, including specific threshold values.
What Every PMS Plan Must Include Under EU MDR
Annex III Section 1(b) specifies the mandatory elements of a PMS plan. Many manufacturers create plans that appear compliant on the surface but lack the specificity needed to withstand Notified Body review, especially around measurable indicators and data analysis methods.
Required Data Sources
The plan must define which sources of information will be actively monitored:
- Serious incident reports
- Non-serious incident records
- Trend data
- Scientific literature (systematic reviews)
- EUDAMED
- Device registries
- User and distributor feedback
- Comparable device data
Simply listing sources is insufficient. The plan must describe how and how often each will be reviewed.
Defined Indicators and Threshold Values
The PMS plan must specify measurable indicators, such as:
- Complaint rates per thousand units sold
- Incident severity distributions
- Return rates by failure mode
Pre-set threshold values trigger action. For example: "If complaint rate exceeds 2% quarterly, initiate root cause investigation and Notified Body notification." These thresholds connect directly to the benefit-risk assessment and must be reviewed and updated over the PMS cycle.
Complaint Investigation and Corrective Action Procedures
The plan must reference or include methods for:
- Investigating complaints
- Analysing field experience
- Initiating CAPA or Field Safety Corrective Actions (FSCAs)
- Traceability tools to identify affected devices
PMCF Plan Integration
The PMCF plan (or a written justification for its exclusion) must be embedded within or formally referenced by the PMS plan. A compliant PMCF plan should contain:
- Objectives (confirm safety/performance, identify unknown side-effects)
- Methods (surveys, registries, literature review, post-market studies)
- Timelines
- Responsible roles
Getting each of these elements right — particularly PMCF objectives and measurable thresholds — is where many manufacturers, especially startups and SMEs, encounter Notified Body pushback. Elexes works with manufacturers to build PMS plans, PMCF plans, and PSURs that meet the specific documentation standards Notified Bodies expect under EU MDR.
PMS Reporting Requirements and Timelines by Device Class
Reporting obligations under EU MDR vary significantly by device risk class — and the differences go beyond update frequency. Each class carries distinct submission routes, report types, and review pathways that manufacturers must build into their PMS systems from the outset.
Reporting Schedule by Device Class
| Device Class | Report Type | Update Frequency | Submission Route |
|---|---|---|---|
| Class I | PMS Report | Updated when necessary | Available to Competent Authority on request |
| Class IIa | PSUR | Every 2 years minimum | Delivered to Notified Body |
| Class IIb (non-implantable) | PSUR | Annually | Delivered to Notified Body |
| Class IIb (implantable) | PSUR | Annually | Submitted via EUDAMED to Notified Body |
| Class III | PSUR | Annually | Submitted via EUDAMED to Notified Body |
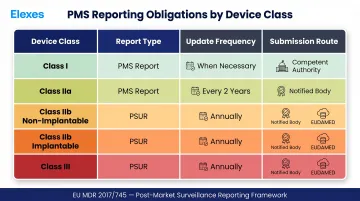
Regardless of class, PMS data collection must remain active and ongoing — not triggered only at reporting intervals.
EUDAMED Implications
For Class IIb implantable and Class III devices, PSURs are submitted via EUDAMED and reviewed by Notified Bodies. Commission Decision (EU) 2025/2371 declared the first four EUDAMED modules functional on 27 November 2025, with mandatory use from 28 May 2026.
Mandatory modules include:
- Actor registration
- UDI/Device registration
- Notified Bodies and Certificates
- Market Surveillance
Not yet mandatory: Vigilance and PMS module. Until this module is functional, manufacturers should submit PSURs directly to their Notified Body — typically via secure file transfer or email — following the submission procedures outlined in MDCG 2022-21.
How PMS Integrates with Your QMS
The Feedback Loop Requirement
Article 83(3) explicitly requires that PMS outputs feed back into at least seven other QMS processes:
- Risk management file updates
- Clinical Evaluation Report (CER) updates
- Labelling and IFU revisions
- CAPA initiation
- Summary of Safety and Clinical Performance (SSCP) updates (Class III and implantable)
- Design and manufacturing information updates
- Identification of options to improve device usability and safety
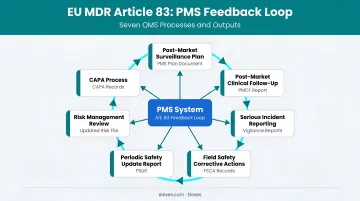
PMS is not a standalone activity. It must actively drive changes across the technical documentation.
The Consistency Challenge Across QMS Records
Because PMS data flows into the CER, risk management file, vigilance records, and PSUR simultaneously, manufacturers must ensure data consistency across all documents. Discrepancies between the CER and PSUR (such as conflicting incident rates or contradictory safety conclusions) are a known Notified Body finding during technical file reviews.
Cross-functional coordination between regulatory, clinical, and quality teams is essential. Elexes supports clients in maintaining this consistency by providing embedded regulatory, clinical, and QMS expertise across all interconnected documents.
QMS Documentation Infrastructure Needed
Manufacturers need defined SOPs for each PMS interface activity, including:
- How PMS findings trigger risk management reviews
- How complaints escalate to vigilance reports
- How PMCF data updates the CER
- How trend analysis initiates CAPA
Without these procedures in place, even a well-written PMS plan will fail during audits.
Common PMS Compliance Gaps and How to Address Them
Most Frequent Gaps Seen During Audits
Common deficiencies include:
- PMS plans lacking defined threshold values and measurable indicators — manufacturers list data sources without specifying how data will be analysed or what triggers action
- PMCF plans missing or replaced by vague justifications — insufficient rationale for why PMCF is not applicable
- PMS data not actively flowing into CER or risk management updates — data collected but not used to drive technical documentation changes
- PSURs not updated on schedule — particularly common among Class IIa manufacturers transitioning from MDD's less prescriptive requirements
- PMS systems treated as documentation exercises rather than active surveillance processes

These patterns are reflected in MDCG guidance documents and regulatory inspection observations. Addressing these gaps starts with knowing exactly where your system falls short.
The Gap Assessment Approach
A structured gap analysis comparing current PMS documentation and processes against EU MDR Articles 83–86 and Annex III requirements is the first step in building or remediating a compliant system. Key areas to benchmark:
- PMS plan completeness
- PMCF status and justification
- Data source coverage
- Indicator definitions
- QMS integration
- Reporting schedules
Prioritisation for Remediation
Manufacturers should prioritise gaps in the order Notified Bodies typically audit them:
- PMS plan and PMCF documentation (technical file elements)
- PSUR completeness (report content and timelines)
- QMS procedure integration (SOPs for data flow)
Class IIb and III manufacturers face the sharpest urgency here — annual PSUR requirements and EUDAMED submission obligations leave little room for delayed remediation.
Conclusion
EU MDR PMS is a continuous, proactive system spanning the entire device lifetime — not a once-a-year reporting task. Manufacturers who treat it as one are systematically exposed to audit findings and market access risk. The shift from MDD to MDR requires structural changes to how manufacturers collect, analyse, and act on post-market data.
To close the gap, manufacturers should take three concrete steps:
- Audit your current PMS system against Annex III requirements and device class reporting obligations
- Build cross-functional QMS integration so PMS data actively drives clinical, risk, and regulatory updates — not just compliance documentation
- Act now: with Notified Body designation timelines exceeding 800 days and limited capacity to clear the legacy certificate backlog, proactive PMS compliance translates directly into market access advantage.
Frequently Asked Questions
What are the post-market surveillance requirements under EU MDR 2017/745?
EU MDR Articles 83–86 require all manufacturers to implement a proactive PMS system, maintain a PMS plan as part of their technical documentation, and produce either a PMS Report (Class I) or a PSUR (Class IIa, IIb, III). The system must be proportionate to device risk class and active throughout the device lifetime.
What does post-market surveillance include?
PMS covers proactive data collection (complaints, incidents, literature, user feedback, registries), systematic analysis, benefit-risk reassessment, CAPA initiation, and PMCF. Outputs feed back into clinical evaluation, risk management, and labelling processes — and trigger vigilance or trend reporting where required.
What is a post-market surveillance plan?
A PMS plan is a mandatory document under Article 84 / Annex III that defines data sources, collection methods, measurable indicators, threshold values, and complaint investigation procedures. It must also include a PMCF plan or justification, and forms part of the technical documentation for all device classes.
What is covered under post-market clinical follow-up (PMCF)?
PMCF involves proactively gathering clinical data after market launch through methods such as post-market studies, patient registries, literature reviews, and user surveys. Its purpose is to confirm ongoing safety and performance, identify previously unknown side effects, and ensure the benefit-risk ratio remains acceptable.
Does ISO 13485 require post-market surveillance?
Yes — ISO 13485:2016 addresses PMS under Clause 8.2 (Monitoring and Measurement), but EU MDR 2017/745 imposes more detailed and prescriptive obligations. Manufacturers CE marking products must satisfy both standards; ISO 13485 certification alone does not fulfill EU MDR PMS requirements.
What is a clinical evaluation report?
A Clinical Evaluation Report (CER) compiles and assesses clinical data to demonstrate a device's safety, performance, and clinical benefit. Under EU MDR Article 61 and Annex XIV, it must be updated continuously using PMS and PMCF outputs — making it a living document tied directly to your post-market surveillance system.


