
Introduction
Achieving CE marking or FDA clearance often feels like crossing the finish line. For medical device manufacturers, it's really just the start of a much longer regulatory journey. The post-market phase is where many companies stumble — particularly when trying to untangle two commonly confused obligations: Post-Market Surveillance (PMS) and Post-Market Clinical Follow-up (PMCF).
These two terms are often used interchangeably, creating confusion that leads to documentation gaps, audit failures, and non-conformities during Notified Body reviews.
Here's the reality: PMS and PMCF are not the same thing. PMS is the comprehensive system that monitors your device's safety, performance, and quality throughout its entire lifecycle. PMCF is a clinical-data-specific component nested within that broader PMS framework. Confusing the two—or worse, treating them as separate, disconnected obligations—exposes manufacturers to serious compliance risks.
This guide clarifies the definitions, structural relationships, key differences, and regulatory documentation requirements for both PMS and PMCF. You'll understand when each applies, how they integrate, and what a defensible post-market strategy actually looks like.
Key Takeaways
- PMS is the overarching lifecycle system for monitoring device safety, performance, and quality — spanning complaints, vigilance, literature, and clinical data.
- PMCF is a clinical subprocess within PMS that proactively collects real-world evidence to confirm benefit-risk after CE marking.
- PMS applies universally to all device classes; PMCF is mandatory for Class III and implantable devices and requires documented justification if not conducted for others.
- Both require formal documentation — PMCF Plan, PMCF Evaluation Report, and either a PMSR (Class I) or PSUR (Class IIa/IIb/III).
- The core difference: PMS casts a wide net reactively; PMCF targets clinical evidence proactively.
PMS vs PMCF: At a Glance
| Dimension | PMS (Post-Market Surveillance) | PMCF (Post-Market Clinical Follow-up) |
|---|---|---|
| Definition | Systematic, ongoing process to monitor device safety, performance, and quality throughout the lifecycle | Proactive, planned collection of real-world clinical data to confirm benefit-risk profile |
| Scope | All quality, performance, and safety data—clinical and non-clinical | Clinical data only—real-world safety and performance evidence |
| Approach | Both reactive (triggered by complaints, incidents) and proactive | Exclusively proactive—planned before data collection begins |
| Data Sources | Complaints, vigilance reports, field actions, user feedback, literature, trend analysis, PMCF data | Registries, post-market clinical studies, literature reviews, surveys, real-world evidence |
| Regulatory Trigger | Mandatory for all device classes under EU MDR Article 83 | Mandatory for Class III/implantable; justified if not conducted for others (Annex XIV Part B) |
| Required Documentation | PMS Plan, PMSR (Class I) or PSUR (Class IIa/IIb/III) | PMCF Plan, PMCF Evaluation Report |
| Device Class Applicability | All classes (I, IIa, IIb, III) | Effectively mandatory for Class III/implantable; expected for IIa/IIb unless justified; justification required for Class I |
| Primary Output | Risk management updates, CAPA, benefit-risk reassessment | Clinical Evaluation Report (CER) updates |

Important clarification: PMCF is not an alternative to PMS—it's a component within it. Vigilance (reactive incident reporting per Articles 87-90) is another distinct component within the PMS ecosystem. Picture PMS as the full monitoring dashboard: PMCF feeds in the clinical-evidence panel, while vigilance handles the incident-alert system. Each has a defined role — gaps in any one of them create compliance exposure across the others.
What is Post-Market Surveillance (PMS)?
Post-Market Surveillance (PMS) is the systematic, ongoing process required by EU MDR Article 83 and equivalent regulations globally. It mandates that manufacturers continuously gather, analyze, and evaluate data on a device's safety, performance, and quality once it reaches the market. Under EU MDR, PMS is mandatory for all device classes and must be "proportionate to the risk class and appropriate for the type of device" — with Article 83(2) requiring active, systematic data gathering across the device's entire lifetime.
Scope of PMS Data Sources
PMS casts a wide net, capturing both clinical and non-clinical signals. Data sources include:
- Complaint handling and customer feedback
- Vigilance reporting (serious incidents, FSCAs, and field safety corrective actions)
- User and distributor feedback
- Trend analysis from quality and service reports
- Post-market clinical studies and PMCF data
- Literature surveillance (specialist journals, databases, registries)
- Publicly available information on similar devices
Per Annex III Section 1(a), the PMS plan must address all these data types and detail how they will be collected, analyzed, and acted upon.
PMS Outputs: PMSR vs. PSUR
PMS data feeds into two distinct reporting formats depending on device class:
Post-Market Surveillance Report (PMSR) – Article 85
- Applies to Class I devices
- Updated "when necessary" (no fixed frequency)
- Summarizes PMS analysis results and rationale for any corrective actions
- Available to competent authorities on request
Periodic Safety Update Report (PSUR) – Article 86
- Applies to Class IIa, IIb, and III devices
- Updated at least every 2 years for Class IIa; at least annually for Class IIb and III
- Must include benefit-risk conclusions, PMCF findings, sales volume, and user population estimates
- Class III and implantable devices: Submitted via EUDAMED to Notified Body for review
- Class IIa/IIb: Available to Notified Body and competent authority on request

Higher-risk devices require more frequent safety documentation — a direct application of the risk-proportionate approach EU MDR mandates throughout.
Integration with Risk Management
PMS data directly informs risk management under ISO 14971:2019 Clause 10. Manufacturers must:
- Evaluate whether new hazards have emerged
- Reassess whether previously acceptable risks remain acceptable
- Determine if the state of the art has changed
- Feed insights back into risk management files and trigger CAPA when necessary
Real-world performance data must be used to continuously reassess the device's benefit-risk ratio. When PMS data reveals a shifting risk profile, manufacturers must update their risk management file — not simply note the finding and move on.
Use Cases of PMS
That closed loop plays out in practice more often than manufacturers expect. Consider a Class IIa diagnostic device where five users in three months reported difficulty interpreting a specific result screen. No adverse events occurred — but trend analysis flagged a usability issue. The manufacturer reviewed the Instructions for Use (IFU), identified ambiguous language, and issued a revised IFU as a preventive action. The manufacturer then documented this update in the next PSUR and referenced it during the Notified Body surveillance audit, demonstrating proactive risk management driven by PMS data.
What is Post-Market Clinical Follow-up (PMCF)?
Post-Market Clinical Follow-up (PMCF) is a planned, systematic clinical data collection process that begins after a device receives CE marking. As defined in EU MDR Annex XIV Part B, PMCF is "a continuous process that updates the clinical evaluation" by generating real-world clinical evidence to confirm that the device's safety and performance holds true across the intended use population over time.
Primary Objectives of PMCF
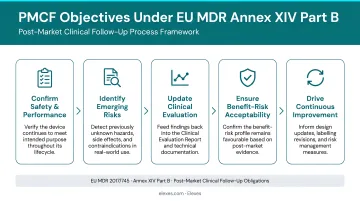
Per EU MDR Annex XIV Part B, PMCF has five core objectives:
- Confirm safety and performance throughout the device's expected lifetime
- Identify previously unknown side-effects and monitor known side-effects and contraindications
- Identify and analyze emergent risks based on factual evidence
- Ensure continued benefit-risk acceptability per Annex I requirements
- Detect systematic misuse or off-label use to verify the intended purpose remains valid
PMCF Methodologies
Manufacturers can choose from multiple approaches, depending on device risk, novelty, and available evidence. Common methods include:
- Registries: Device-specific registries or evaluation of public registries
- Post-market clinical studies: Prospective follow-up of pre-market patients, new observational studies, or retrospective analyses
- Systematic literature reviews: Screening of scientific literature and clinical databases
- Real-world evidence (RWE): Analysis of real-world data from reliable sources
- User surveys: Planned data collection from healthcare professionals or patients
- Case reports: Review to identify misuse or off-label use patterns
The choice of method must be justified based on device classification, risk profile, and the strength of existing clinical evidence.
Required PMCF Documents
Under EU MDR, two core documents are mandatory:
PMCF Plan (MDCG 2020-7)
- Outlines objectives, methods, and endpoints
- References the Clinical Evaluation Report (CER) and risk management
- Includes justified timelines and acceptance criteria
- Must be part of the technical documentation
PMCF Evaluation Report (MDCG 2020-8)
- Summarizes findings and their impact on the CER
- Must be part of the CER and technical documentation
- Conclusions feed directly into clinical evaluation and risk management
Use Cases of PMCF
Orthopedic implant (Class IIb): A manufacturer launches a 3-year registry study to monitor long-term performance outcomes, capturing revision rates, patient-reported outcomes, and adverse event incidence. The registry data feeds annually into the PMCF Evaluation Report, which updates the CER and informs the PSUR.
Software as a Medical Device (SaMD): A diagnostic algorithm developer uses real-world performance data from deployed devices to validate accuracy in diverse patient populations. This RWE confirms the algorithm's benefit-risk profile across demographic groups not fully represented in pre-market validation studies.
Established diagnostic device (Class IIa): A manufacturer conducts a systematic literature review as a justified alternative to a new PMCF study, demonstrating that the device type is well-characterized and no new safety signals have emerged in the literature over the past two years.
Elexes provides end-to-end support in building PMCF plans and evaluation reports, including literature-based PMCF, study design, and CER integration—helping manufacturers build PMCF as a living evidence-generation system that evolves with the device's real-world use.
PMS vs PMCF: Key Differences Explained
Difference 1 — Scope: Ecosystem vs. Clinical Subset
PMS covers the entire post-market safety and performance monitoring ecosystem—including non-clinical data like complaints, service reports, and vigilance. PMCF is exclusively clinical, counting only data that demonstrates real-world clinical safety and performance.
Analogy: PMS is the full health monitoring dashboard; PMCF is one vital-signs panel on that dashboard.
Difference 2 — Approach: Reactive vs. Proactive
PMS includes both reactive processes (triggered by adverse events, complaints, or incident reports) and proactive ones. PMCF is inherently proactive—it must be planned before data collection begins and cannot be initiated in response to an incident. EU MDR Annex XIV Part B requires PMCF to be systematically planned with defined objectives, methods, and timelines.
Difference 3 — Regulatory Driver and Output
PMS outputs flow into the PMSR or PSUR and inform risk management files. PMCF outputs flow specifically into the Clinical Evaluation Report (CER). This distinction matters because the CER is a living document that Notified Bodies scrutinize closely—outdated or absent PMCF evidence is a top reason for CER rejection during MDR certification.
Difference 4 — Relationship to Vigilance
Vigilance (mandatory reporting of serious incidents and FSCAs per Articles 87-90) is a distinct third component within PMS—separate from PMCF. Vigilance is reactive and event-triggered, with strict reporting timelines (2–15 days depending on severity). PMCF is proactive and planned.
Example: An X-ray device manufacturer reports no serious incidents under vigilance over two years. A systematic literature review conducted as part of PMCF reveals that acceptable radiation exposure thresholds have decreased globally based on new research. This state-of-the-art shift requires updating the device's risk management file and CER. Vigilance data alone would never surface it.
Difference 5 — Device Class Applicability
PMS is universal—it applies to all device classes in all markets. PMCF intensity varies:
- Class III and implantable devices: PMCF is mandatory. The PSUR must include "main findings of the PMCF," and manufacturers must submit these PSURs annually to the Notified Body via EUDAMED.
- Class IIa and IIb: PMCF is strongly expected unless the manufacturer provides a documented scientific justification.
- Class I: A PMCF study may not be required, but the justification must appear in the technical file.
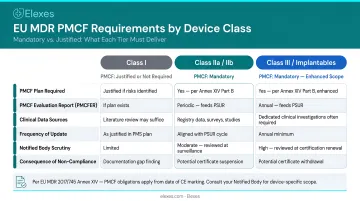
Per Annex III Section 1(b), the PMS plan must include "a PMCF plan as referred to in Part B of Annex XIV, or a justification as to why a PMCF is not applicable."
The five differences above are easier to compare side by side:
| Dimension | PMS | PMCF |
|---|---|---|
| Scope | All post-market data (clinical + non-clinical) | Clinical data only |
| Approach | Reactive and proactive | Proactive and planned |
| Primary Output | PMSR / PSUR, risk management file | Clinical Evaluation Report (CER) |
| Vigilance Link | Includes vigilance as a sub-component | Separate from vigilance; not event-triggered |
| Device Class | Applies to all classes | Mandatory for Class III; expected for IIa/IIb |
When is PMCF Required Under EU MDR?
PMCF requirements are driven by device classification and risk profile. Here's the breakdown:
Class III and implantable devices:
- PMCF is mandatory
- The PSUR must include PMCF findings and is submitted annually via EUDAMED
- Notified Bodies review PMCF data as part of surveillance audits
Class IIa and IIb:
- PMCF is expected
- Justification for not conducting PMCF must be robust and documented
- The PSUR (updated every 2 years for IIa, annually for IIb) must still address PMCF applicability
Class I:
- A documented justification for not conducting PMCF must be included in the PMS plan within the technical documentation
Per MDCG 2020-7, Notified Bodies assess the manufacturer's "justification in relation to non-performance of PMCF," placing the burden of proof on the manufacturer.
Triggers for New or Expanded PMCF
PMCF is not a one-time study—it's a living process. New or expanded PMCF activities may be triggered by:
- New safety signals from PMS or vigilance data
- Significant changes in device design or intended use
- New state-of-the-art evidence that challenges the current benefit-risk profile
- Notified Body feedback during surveillance audits
- Unanswered questions about long-term safety or performance in specific populations
Understanding these triggers matters especially for dual-market manufacturers, where EU MDR obligations and FDA requirements follow very different logic.
EU MDR vs. FDA Post-Market Requirements
The EU MDR and FDA frameworks for post-market clinical data collection are different in structure and intent:
| Feature | EU MDR PMCF | FDA Section 522 |
|---|---|---|
| Mandate Type | Universal expectation for all classes (justify if not done) | Discretionary—FDA issues specific orders |
| Initiation | Manufacturer-driven, continuous | Regulator-triggered based on specific concerns |
| Trigger Conditions | Inherent in MDR compliance | Four specific triggers: (1) failure likely to cause serious harm, (2) significant pediatric use, (3) implanted >1 year, (4) life-sustaining/supporting outside professional facility |
| Device Classes | All classes (I, IIa, IIb, III) | Class II and III only |
| Scope | Lifecycle-long, proactive | Targeted surveillance questions |

The key difference: EU MDR treats post-market clinical data collection as a standing manufacturer obligation built into the QMS. FDA deploys it as a targeted tool when specific risk thresholds are met. Manufacturers operating in both markets need two distinct compliance frameworks to reflect that difference.
Conclusion
PMS and PMCF are not competing obligations—they are complementary layers of a compliant post-market system. PMS is the broad monitoring framework that captures all safety, performance, and quality signals throughout the device lifecycle. PMCF is the clinical-data-specific component within PMS, proactively generating real-world evidence to keep the Clinical Evaluation Report current and the benefit-risk profile valid.
Knowing which data sources belong where, what outputs each system generates, and when regulatory triggers apply will prevent documentation gaps and audit failures. PMCF is an ongoing evidence-generation commitment — not a one-time clinical exercise — and that distinction directly sustains the CER and CE marking.
Manufacturers who treat PMS and PMCF as integrated, living systems rather than periodic compliance checkboxes consistently perform better in Notified Body surveillance audits and respond faster to safety signals or market changes.
Elexes works with medical device companies to build and maintain compliant PMS and PMCF systems across EU MDR, FDA, and other global frameworks — from initial regulatory strategy through ongoing post-market obligations.
Frequently Asked Questions
What is PMS and PMCF?
PMS (Post-Market Surveillance) is the comprehensive post-market monitoring system required for all device classes under EU MDR Article 83. PMCF (Post-Market Clinical Follow-up) is a clinical-data-specific subprocess within PMS that proactively collects real-world clinical evidence after CE marking. Both are required, but they serve distinct roles in the post-market compliance framework.
What is PMCF in clinical trials?
PMCF is not a traditional pre-market clinical trial—it is a post-market clinical evidence collection process that may use study designs similar to clinical trials (such as registries and prospective observational studies). PMCF occurs after CE marking to confirm real-world safety and performance in the intended use population over time.
Is PMCF required for Class I?
PMCF studies are not mandatory for Class I devices, but manufacturers must include a documented justification in the technical file explaining why a PMCF study is not applicable. The absence of a plan must be actively justified under Annex III Section 1(b) — it cannot simply be omitted.
Is PMCF part of PMS?
Yes. PMCF is a subset of PMS—specifically the clinical data collection component. PMS is the broader system encompassing complaints, vigilance, literature surveillance, and PMCF. PMCF focuses on generating real-world clinical evidence to update the CER.
What documents are required for PMCF under EU MDR?
Two documents are required: the PMCF Plan (outlining objectives, methods, and timelines per MDCG 2020-7) and the PMCF Evaluation Report (summarizing findings and their impact on the CER per MDCG 2020-8). Both must be included in the device's technical documentation per EU MDR Annex XIV Part B.
What is the difference between a PMSR and a PSUR?
The Post-Market Surveillance Report (PMSR) applies to Class I devices and is updated "when necessary" without a fixed frequency. The Periodic Safety Update Report (PSUR) applies to Class IIa, IIb, and III devices—with Class IIa updated at least every 2 years and Class IIb/III updated at least annually. Both summarize PMS and PMCF findings.


