
Introduction
Design flaws that reach patients don't just cause harm — they trigger recalls, FDA enforcement actions, and in some cases, market withdrawals. 21 CFR 820.30, the FDA's design controls regulation under the Quality System Regulation (QSR), exists precisely to prevent that outcome. It requires manufacturers to establish documented procedures that systematically translate design intent into verified, safe devices.
This matters because inadequate design controls remain a leading cause of device recalls and FDA enforcement actions. According to the FDA's landmark study, 44% of voluntary device recalls between 1983 and 1989 could have been prevented by adequate design controls — a finding that directly shaped the modern QSR framework. Even today, design control deficiencies rank among the most frequently cited observations on FDA Form 483s.
The regulation applies to all Class II and Class III device manufacturers, plus certain Class I devices — including software-automated systems. Whether you're building a quality system from scratch or preparing for an FDA inspection, understanding each of 820.30's 10 elements is the foundation for staying compliant and avoiding enforcement risk.
Key Takeaways
- 21 CFR 820.30 requires manufacturers to control the design process through documented, reviewable procedures
- It contains 10 specific requirements—from design planning to the Design History File (DHF)—each must be established, maintained, and documented
- Applies to all Class II and Class III devices, plus specific Class I devices including software-automated devices
- Verification confirms output meets input; validation confirms the device meets user needs — the two are distinct requirements, not interchangeable
- Under the QMSR update (effective February 2026), 820.30 is reserved; design controls now fall under ISO 13485 Clause 7.3
What Is 21 CFR 820.30 Design Controls?
21 CFR 820.30 is the Subpart C section of the FDA's Quality System Regulation (21 CFR Part 820), establishing a structured framework of quality practices that must be integrated into device design and development. Its purpose is to ensure the final device meets user needs, intended uses, and specified safety and performance requirements.
Understanding how design controls differ from GMP is essential for compliance. Good Manufacturing Practices govern production quality — the how of building a device. Design controls govern what gets built, applied before the device ever reaches production. That distinction is why the QSR elevated standards beyond prior cGMP frameworks: most device failures trace back to flawed design, not flawed manufacturing.
The key differences at a glance:
- GMP — governs manufacturing processes and production quality
- Design controls — govern the design process before production begins
- Scope — design controls address the root cause of device failures that GMP cannot catch
Design controls aren't a one-time activity. They apply across the full device lifecycle: premarket (once a device moves beyond feasibility or proof-of-concept) and postmarket (for managing design changes and sustaining compliance). Every design change triggers design control obligations anew.
The 10 Requirements: How 21 CFR 820.30 Works
820.30 contains 10 subsections (a through j), each addressing a specific phase or function within design and development. Collectively they form a closed-loop system: inputs drive outputs, outputs are reviewed and tested, and all decisions are documented in a traceable record.
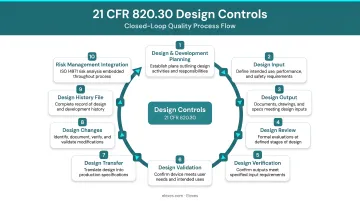
Design Planning, Input, and Output
Design and Development Planning (§820.30(b)): Manufacturers must establish and maintain a documented plan that describes all design activities, identifies cross-functional interfaces, and assigns responsibilities. This plan must be updated and approved as the design evolves. It keeps teams aligned throughout development and should be treated as a living document, not something filed once and ignored.
Design Input (§820.30(c)) and Design Output (§820.30(d)): These two requirements work in tandem and must be documented separately:
- Design inputs are documented physical and performance requirements derived from user needs—what the device must do
- Design outputs are technical results—drawings, specifications, software code, test protocols—that must demonstrably satisfy those inputs
Both must be documented and approved by designated individuals. Vague inputs lead to unverifiable outputs—one of the most common and costly FDA observations.
Design Review, Verification, and Validation
Design Review (§820.30(e)): Formal, documented reviews must occur at appropriate stages of development and include representatives from all relevant functions plus an independent reviewer not directly responsible for the stage under review. Results must be recorded in the DHF. The purpose is to surface problems early, while corrections are still low-cost.
Design Verification (§820.30(f)) and Design Validation (§820.30(g)): These are frequently confused, and FDA auditors consistently cite this misunderstanding:
- Verification: "Did we build the product correctly?" Confirms output meets input requirements through objective evidence—lab tests, inspections, dimensional checks, software unit tests
- Validation: "Did we build the correct product?" Confirms the device meets defined user needs and intended uses under actual or simulated conditions on production units
Both are required. Neither substitutes for the other. Per FDA guidance, validation must include software validation and risk analysis where appropriate.

Design Transfer, Changes, and the Design History File
Design Transfer (§820.30(h)): The critical bridge from design to production. Procedures must ensure the finalized design is correctly translated into production specifications—preserving design intent and confirming manufacturing feasibility. Leaving design transfer to final stages of development is a common, costly mistake.
Design Changes (§820.30(i)): All design changes must follow a documented process before implementation:
- Identify and document the change
- Verify or validate as appropriate
- Review and approve by designated personnel
- Assess whether the change triggers a new premarket submission
Design History File (§820.30(j)): The DHF is the cumulative record of the entire design history for each device type. It must contain or reference all records demonstrating the design was developed per the approved plan and 820.30 requirements. Incomplete DHFs are among the most cited deficiencies in FDA inspections.
Who Must Comply With 21 CFR 820.30?
All Class II and Class III device manufacturers must comply with 820.30 in full. Most Class I devices are exempt, but these Class I devices are explicitly subject to design controls:
- Devices automated with computer software
- Tracheobronchial suction catheters
- Surgeon's gloves
- Protective restraints
- Manual radionuclide applicator systems
- Radionuclide teletherapy sources
Two compliance details catch manufacturers off guard:
- Class I "exempt" status has limits. Exemption from 820.30 doesn't eliminate all QMS obligations — labeling accuracy, record-keeping, and general quality system requirements may still apply. Conduct a device-specific review rather than assuming full exemption.
- Scope extends beyond the original designer. Contract manufacturers, specification developers, and remanufacturers are all subject to design controls. This is a common source of compliance gaps in outsourced development arrangements.
Key Factors That Shape Effective Design Control Implementation
Critical inputs affecting design control outcomes:
- Completeness and traceability of design inputs — vague or undocumented inputs cascade into unverifiable outputs
- Cross-functional team involvement — engineering, clinical, regulatory, and quality functions must be engaged from the start
- Risk management integration — ISO 14971 risk analysis should be embedded in design inputs and validation, not treated as a standalone activity at the end
- Software validation requirements (IEC 62304) — SaMD and software-integrated devices carry additional validation obligations that must be planned early
Documentation discipline is a structural requirement, not a procedural formality. Every review, approval, verification test, and validation report must be date-stamped and signed by identified individuals. Incomplete or retroactively created records are a primary source of Form 483 observations and warning letters.

Companies working across global markets should also align design control documentation with ISO 13485 Clause 7.3, since the FDA's QMSR now requires compliance with that standard. Building a system that satisfies both FDA and international requirements from the outset is far less costly than retrofitting it later. Elexes brings ISO 13485 and ISO 14971 expertise across 250+ device projects to help manufacturers do exactly that.
Common Mistakes and Misconceptions Under 21 CFR 820.30
These five patterns appear repeatedly in FDA 483 observations and warning letters. Recognizing them early can prevent costly remediation downstream.
Treating Design Controls as Documentation, Not Infrastructure
The most damaging misconception in design control compliance. Manufacturers who retrofit documentation at the end of development—rather than building controls into the process—produce DHFs that cannot demonstrate real compliance. This immediately raises red flags during audits and premarket submissions.
Conflating Verification with Validation
Teams often perform one instead of the other, or treat them as interchangeable. The distinction matters:
- Verification: Objective evidence that design output meets design input (lab tests, dimensional checks, analysis)
- Validation: Evidence the device satisfies actual user needs (clinical studies, simulated-use testing, human factors evaluations)
Both are required. Neither substitutes for the other.
DHF Incompleteness
820.30 requires the DHF to contain or reference all records demonstrating development followed the approved plan. Yet many manufacturers maintain incomplete DHFs missing approval signatures, change history, or validation rationale—one of the most cited deficiencies in FDA 483 observations.
The downstream consequences extend beyond audits. A deficient DHF becomes a remediation liability in M&A transactions, directly affecting commercial value.
Treating Design Transfer as a Final-Stage Handoff
Manufacturers often finalize production specifications only at the end of development. When manufacturing issues surface late, the result is costly design iteration loops that could have been avoided. Design transfer should be considered from early development stages, particularly for devices with complex manufacturing tolerances or supply chain dependencies.
Misunderstanding Software Device Scope
Many SaMD manufacturers incorrectly assume that because their device is "software only," physical device design control requirements don't apply. In fact, devices automated with computer software are explicitly named in 820.30(a)(2) as Class I devices subject to design controls.
IEC 62304 software lifecycle requirements must be integrated into the design control process—not treated as a separate compliance track.
21 CFR 820.30 and the QMSR Transition: What's Changing?
In February 2024, the FDA published the Quality Management System Regulation (QMSR) final rule, effective February 2, 2026. Under the QMSR, § 820.30 is reserved — no longer active regulatory text.
Design control requirements now live under § 820.10(c), which mandates compliance with ISO 13485:2016 Clause 7.3 (Design and Development) and its subclauses.
What Stays the Same vs. What Changes
The core design control elements — planning, input, output, review, verification, validation, transfer, changes, and the DHF equivalent — are all preserved in ISO 13485 Clause 7.3. Key differences include:
- Stronger explicit emphasis on risk-based thinking throughout design inputs (not just at validation)
- Harmonized terminology with international standards
- Removal of some QSR-specific procedural requirements now subsumed into the ISO standard

Manufacturers already ISO 13485 certified should find the transition more straightforward.
Transition Strategy
Start with a gap analysis comparing your existing 820.30-based design control procedures against ISO 13485 Clause 7.3 requirements. From there:
- Companies still operating under legacy QSR processes after February 2, 2026 risk non-compliance
- FDA's QMSR FAQ covers implementation timelines and agency expectations
The sooner you map your current procedures to the new standard, the less disruptive the transition will be.
Frequently Asked Questions
What is the FDA 21 CFR 820.30 design controls?
21 CFR 820.30 is the FDA regulation under the Quality System Regulation (21 CFR Part 820) requiring medical device manufacturers to establish and maintain documented procedures controlling the design and development process, ensuring devices meet user needs, intended uses, and safety requirements.
Does ISO 13485 cover design controls?
Yes, ISO 13485:2016 Clause 7.3 covers design and development controls. Under the FDA's new QMSR (effective February 2026), compliance with Clause 7.3 and its subclauses now serves as the basis for meeting FDA design control requirements, replacing the legacy 21 CFR 820.30 text.
What is DHF vs DMR vs DHR?
The Design History File (DHF) documents the design development history of a device; the Device Master Record (DMR) contains instructions and specifications for manufacturing the device; and the Device History Record (DHR) is the production record for each batch or unit manufactured—three distinct files serving different compliance purposes.
What is DFM in medical devices?
Design for Manufacturing (DFM) is the practice of designing a device with manufacturability in mind from early development stages, ensuring design outputs can be reliably and consistently produced at scale. It's directly relevant to the Design Transfer requirement under 820.30(h).
What is a design control document?
Design control documents are the records required under 820.30: the design plan, input and output specifications, review minutes, verification and validation protocols, and change records. These documents are compiled into the Design History File (DHF) to demonstrate that development followed the approved plan.
What is 483 in FDA audit?
A Form 483 is an FDA observation document issued at the close of an inspection when investigators identify conditions that may violate FDA regulations. Design control deficiencies — incomplete DHFs, missing approval signatures, and undocumented design changes — rank among the most frequently cited observations.


