
Introduction
FDA medical device labeling violations consistently rank among the top Form 483 citations issued to device manufacturers. According to FDA's publicly available inspection observation data, labeling control failures under 21 CFR 820.120 appear repeatedly across fiscal years as one of the most frequently cited device observations, alongside design control deficiencies. These citations carry serious consequences: delayed 510(k) clearance, warning letters, product recalls, and in severe cases, consent decrees that halt manufacturing operations entirely.
FDA enforcement actions consistently cite labeling violations including misbranding under FFDCA Section 502, inadequate directions for use, omission of required UDI elements, and off-label promotional claims. The volume of labeling-related warning letters has trended upward in recent years, reflecting both increased FDA scrutiny and the expanding scope of what qualifies as a regulated label.
That scope is broader than most manufacturers expect. FDA's definition of "labeling" extends well beyond the physical product label to include Instructions for Use (IFU), promotional materials, websites, training documents, and software user interfaces.
This guide walks through the full labeling compliance picture: statutory definitions, required content under 21 CFR Part 801, UDI requirements, quality system controls under 21 CFR 820.120, device-class-specific rules, and the compliance gaps most commonly cited in Form 483 observations.
Key Takeaways
- Medical device "labeling" includes far more than the sticker on the box—it covers IFUs, packaging inserts, promotional materials, websites, and software interfaces.
- 21 CFR Part 801 governs all US device labeling: manufacturer identification, directions for use, warnings, and prominence requirements.
- The UDI system (21 CFR Part 801 Subpart B) mandates a machine-readable and human-readable identifier on most device labels, with GUDID database submission required.
- 21 CFR 820.120 requires documented QMS controls for labeling inspection, storage, and Device History Record (DHR) entries.
- Top violations: label mix-ups, missing UDI or expiration dates, off-label promotional claims, and undocumented labeling inspections.
What Is Medical Device Labeling? Label vs. Labeling Defined
The FDA draws a sharp legal distinction between "label" and "labeling" — and getting this wrong is one of the most common compliance gaps manufacturers face. Both terms are defined under Section 201 of the Federal Food, Drug, and Cosmetic Act (FFDCA), and each carries different regulatory obligations:
Label (21 U.S.C. 321(k)): Written, printed, or graphic matter on the immediate container of a device. If there is an outer retail package, the label information must also appear on that outer container or be easily legible through it.
Labeling (21 U.S.C. 321(m)): All labels and other written, printed, or graphic matter (1) upon the device or its containers or wrappers, or (2) "accompanying" the device. FDA interprets "accompanying" broadly to include promotional brochures, training materials, website content, and even digital displays shown at trade shows or sent via email.
What Falls Under FDA's Definition of Labeling
The following are all considered labeling and must comply with 21 CFR Part 801:
- Physical product labels affixed to the device
- Packaging and outer cartons
- Instructions for Use (IFU) and user manuals
- Package inserts
- Software user interface screens (including splash screens, error messages, and help menus)
- Promotional materials, sales brochures, and website content describing the device
- Training videos and educational materials distributed to users

Every written communication about a device — whether on the box or on a sales rep's tablet — is subject to FDA labeling regulations. Off-label claims in promotional materials can trigger misbranding charges even if the physical label itself is fully compliant.
ISO 13485:2016 and Labeling
While FFDCA defines labeling's legal scope, ISO 13485:2016 governs how labeling is controlled within your quality management system — specifically through Clause 7.5.1 (Control of production and service provision) and Clause 4.2.3 (Medical device file). ISO 13485 requires that labeling include:
- Device identification (name, model, serial number)
- Technical descriptions
- Intended purpose and proper use instructions
- Warnings, contraindications, and precautions
The February 2026 transition to the Quality Management System Regulation (QMSR) incorporates ISO 13485 by reference while adding FDA-specific supplemental requirements for labeling inspection and DHR documentation.
Required Label Elements Under 21 CFR Part 801
Name and Place of Business (21 CFR 801.1)
Every device label must include the corporate name and full street address (city, state, ZIP) of the manufacturer, packer, or distributor. If the company whose name appears on the label is not the manufacturer, a qualifying phrase is required:
- "Manufactured for [Company Name]"
- "Distributed by [Company Name]"
- Any other wording that accurately describes the relationship
Adequate Directions for Use (21 CFR 801.5)
"Adequate directions for use" means instructions a layperson can follow to use the device safely and for its intended purposes. The directions must cover:
- Conditions, purposes, and uses for which the device is intended
- Dosage or frequency of use
- Route of administration or method of application
- Duration of treatment
- Preparation or assembly steps
Exemption for Prescription Devices: Devices restricted to use by a licensed practitioner and bearing "Rx only" (per 21 CFR 801.109) are exempt from layperson directions.
However, exemption is not a free pass. These devices must still carry professional-use labeling that includes clinical information, contraindications, adverse events, and hazards.
Mandatory Content Elements
Every medical device label must include:
- Device name and intended use statement
- Indications, contraindications, precautions, and warnings
- Manufacture and expiration dates formatted as YYYY-MM-DD per 21 CFR 801.18 (e.g., 2025-03-15)
- Lot or serial number for traceability
- Manufacturer contact information (name, address)
- Storage instructions if special conditions apply
- "STERILE" statement if applicable

Once you know what to include, placement and legibility determine whether those elements actually satisfy 21 CFR 801.15.
Prominence of Required Statements (21 CFR 801.15)
Required information must be:
- Conspicuous and legible: Adequate font size, sufficient contrast, no overcrowding with non-required content
- Appropriately placed: On the principal display panel where the consumer can see it
- In English: If any foreign language appears on the label, all required information must also appear in English
Use of Symbols (21 CFR 801.15(c))
FDA recognizes ISO 15223-1:2021 for standardized medical device symbols. When symbols appear without adjacent explanatory text, a glossary or symbol key must accompany the device. Common symbols include:
- Manufacturer symbol
- Date of manufacture
- Use-by date
- Batch code
- Consult instructions for use
- Caution
Subpart D Exemptions (21 CFR 801.109–801.125)
Certain device categories may qualify for partial exemptions from adequate directions requirements:
- Prescription devices (21 CFR 801.109)
- Devices for use by licensed practitioners (21 CFR 801.110)
- In vitro diagnostic products (21 CFR 809.10)
- Laboratory/research-use-only devices
Important: These exemptions have specific qualifying conditions — misclassifying a device as exempt is a compliance violation in itself. Verify eligibility carefully; residual labeling obligations still apply even when an exemption is granted.
UDI Requirements Under 21 CFR Part 801 Subpart B
UDI Mandate (21 CFR 801.20)
Every medical device label and package must bear a Unique Device Identifier (UDI) consisting of two components:
- Device Identifier (DI): Fixed portion that identifies the specific version, model, and labeler — constant across all units of that device version.
- Production Identifier (PI): Variable portion encoding lot or batch number, serial number, expiration date, manufacturing date, or distinct identification code (for HCT/Ps).
The PI is required whenever any of these elements are present on the label.
Once you know what the UDI must contain, the next question is how it must appear on the label.
Format Requirements (21 CFR 801.40)
The UDI must appear in two forms on the label:
- Human-readable plain text — legible by sight without any equipment
- AIDC technology — barcode, RFID, or another machine-readable format

Direct Marking (21 CFR 801.45): Reusable devices intended for reprocessing between uses must carry a permanent UDI marking directly on the device itself. Exceptions apply only where marking would compromise safety, effectiveness, or is not technologically feasible.
UDI Exemptions (21 CFR 801.30)
Twelve device categories are exempt from UDI requirements under 21 CFR 801.30, including:
- Class I devices exempt from GMP requirements under 21 CFR Part 820
- Investigational devices and clinical trial devices under 21 CFR Part 812
- Custom devices (per 21 CFR 812.3(b))
- Export-only devices and veterinary devices
- Research, teaching, or chemical analysis devices (non-clinical use)
- Strategic National Stockpile devices
- Bulk-packaged single-use devices (outer package still requires UDI)
- NDC-bearing combination products
- Shipping containers
For the complete list of exemptions, refer directly to 21 CFR 801.30.
Class I Production Identifier Exemption: Even if a Class I device label includes a lot or serial number, a production identifier (PI) is not required under 21 CFR 801.30(d).
GUDID Submission Requirements
Device labelers must submit identification information to FDA's Global Unique Device Identification Database (GUDID) before first commercial distribution in the United States. Key submission obligations:
- Submit all required device data elements before initial US market distribution
- Submit updates within 60 days of any changes to device labeling, intended use, or other GUDID data elements
- Maintain GUDID records to reflect the current, commercially distributed version of the device
Stand-Alone Software (SaMD) Provision (21 CFR 801.50)
Software not distributed in packaged form may display its UDI through:
- A UDI statement at each software startup, or
- An "About" menu command or similar interface element
The production identifier for SaMD must include the software version.
Labeling Controls Under FDA QSR (21 CFR 820.120)
Overarching Requirement
Under 21 CFR 820.120, manufacturers must establish and maintain documented procedures to control all labeling activities throughout the product lifecycle. Labeling compliance spans both content accuracy and quality system discipline.
Transition to QMSR: That framework is now changing. The Quality Management System Regulation (QMSR) final rule, published February 2, 2024, replaces 21 CFR 820.120 with ISO 13485-aligned requirements effective February 2, 2026. The new regulation references ISO 13485 Clause 7.5.1 while adding FDA-specific supplemental requirements for labeling inspection and documentation.
Labeling Integrity and Storage
Label Integrity: Labels must remain legible and affixed under all storage, distribution, and use conditions. Faded, detached, or smeared labels constitute a violation.
Storage controls exist to prevent mix-ups between similar SKUs and ensure proper identification at every stage. Best practices include:
- Physical segregation of labels for different products, lots, or versions
- Access controls (restricted areas, authorized personnel only)
- Clear identification systems (bin labels, shelf markers, color coding)
- First-in, first-out (FIFO) inventory management
Labeling Inspection and Documentation
Before labeling is released for use, a designated individual must inspect it for accuracy. That inspection must cover:
- UDI formatting (both human-readable and AIDC)
- Expiration dates and manufacture dates (YYYY-MM-DD format)
- Control numbers (lot or serial)
- Storage instructions and warnings
- Manufacturer name and address
- Correct product name and intended use
Every inspection must be captured in the Device History Record (DHR). Required documentation includes:
- Date of inspection
- Signature or electronic approval of the inspector
- Acceptance criteria and results
- Corrective actions for any non-conformances

Missing DHR documentation is one of the most cited labeling-related Form 483 observations.
Labeling Operations and Change Control
Positive release checks ensure the correct label is applied to the correct device or lot before product ships. Steps typically include:
- Line clearance procedures before label runs
- Scanning verification systems
- Sampling and visual inspection during production
- Reconciliation of label quantities issued versus used
When labels change, a formal change management process applies. Any labeling update must go through:
- Review and approval by regulatory, quality, and clinical functions
- Re-validation of labeling procedures if process changes
- Regulatory impact assessment (does the change require a new 510(k) or PMA supplement?)
- Documentation of change rationale, approval, and implementation date
Device-Class-Specific Labeling Considerations
Prescription Device Labeling (Rx Devices)
Under 21 CFR 801.109(b)(1), prescription devices must display one of the following statements:
- "Rx only" or "℞ only"
- "Caution: Federal law restricts this device to sale by or on the order of a [physician/dentist/veterinarian]"
Beyond the required statement, prescription device labeling must also include:
- Detailed clinical use information
- Indications and contraindications
- Adverse event information
- Hazards and precautions for practitioners
- Instructions for use, maintenance, and reprocessing
OTC (Over-the-Counter) Device Labeling
OTC device labeling is governed by 21 CFR Part 801 Subpart C and must include:
Principal Display Panel (21 CFR 801.60):
- Statement of identity (21 CFR 801.61): The name of the device
- Net quantity of contents (21 CFR 801.62): The amount in the package
Directions for use must be written in plain language that an average user can follow without professional supervision. FDA recommends usability testing to confirm that target users can correctly interpret and act on the labeling.
Software as a Medical Device (SaMD) and Digital Devices
SaMD introduces labeling requirements that go beyond traditional physical devices. Three areas demand particular attention: electronic labeling, UDI display, and cybersecurity disclosures.
Electronic labeling (eIFU): FDA permits electronic labeling for prescription devices used in healthcare facilities under Section 206 of the Medical Device User Fee and Modernization Act (MDUFMA). The applicable guidance is "Electronic Labeling for Prescription Devices Intended for Use in Health Care Facilities" (Guidance #G03-1).
UDI display for SaMD (21 CFR 801.50): Stand-alone software must show its UDI at startup or through a menu command. The production identifier must include the software version.
Cybersecurity labeling: FDA's September 2023 final guidance on cybersecurity in medical devices applies to all software-containing devices. Labeling should address:
- Cybersecurity risk management approach
- Security features and user authentication requirements
- Software update mechanisms
- Known vulnerabilities and mitigation strategies
Common FDA Medical Device Labeling Compliance Pitfalls
Top Labeling Violations Seen in Form 483 Observations and Warning Letters
Label Mix-Ups: Inadequate storage controls or SKU confusion leading to incorrect labels applied to devices. This violates 21 CFR 820.120(c) and can result in field actions.
Omission of Required Information: Missing UDI, expiration dates, lot numbers, or manufacturer address. These are per-se violations of 21 CFR Part 801 and FFDCA Section 502(f).
Off-Label Promotional Claims: Marketing materials, websites, or sales presentations making claims that exceed the cleared/approved intended use. Under FFDCA Section 201(m), FDA treats these materials as "labeling" — triggering misbranding charges.
Unapproved Symbols Without Glossary: Using ISO 15223-1 symbols on labels without providing an adjacent explanatory text or symbol key violates 21 CFR 801.15.
Failure to Document Labeling Inspections in the DHR: Missing signatures, dates, or acceptance criteria in DHR records is a recurring 21 CFR 820.120(b) citation.
Incorrect Date Format: Using MM-DD-YYYY or DD/MM/YYYY instead of the mandated YYYY-MM-DD format under 21 CFR 801.18.

Building a Proactive Labeling Compliance Program
Establish Written SOPs: Develop standard operating procedures for labeling development, review, approval, storage, inspection, and change control.
Cross-Functional Review Before Label Approval — involve all relevant stakeholders:
- Regulatory affairs (compliance with 21 CFR Part 801, UDI, exemptions)
- Clinical/medical affairs (accuracy of indications, contraindications, warnings)
- Legal (promotional claims, intellectual property)
- Quality (DHR documentation, labeling inspection procedures)
Usability Testing: Patient-facing labeling — OTC and home-use devices — requires human factors testing to confirm users can safely understand and follow instructions.
Regular Internal Audits: Audit labeling storage areas, DHR records, and change control logs quarterly or semi-annually to catch issues before FDA inspections.
Multi-Market Labeling Complexity
A solid internal compliance program is the foundation — but manufacturers launching in multiple markets face an additional layer of compounding requirements across every framework simultaneously:
| Requirement | FDA (21 CFR 801) | EU MDR (Annex I Ch. III) | Health Canada (SOR/98-282) |
|---|---|---|---|
| Language | English (bilingual if foreign language present) | Official language(s) of each Member State | English AND French (bilingual) |
| Unique ID | UDI (DI + PI) | UDI-DI per Article 27 | Device identifier |
| Date Format | YYYY-MM-DD mandated | Per harmonized standards | ISO 8601 recommended |
| Electronic Labeling | Permitted for Rx devices in healthcare facilities | Permitted under specific conditions (Reg. 2021/2226) | Permitted for professional-use devices |
| Symbols Standard | ISO 15223-1:2021 recognized | ISO 15223-1 mandated | ISO 15223 referenced |
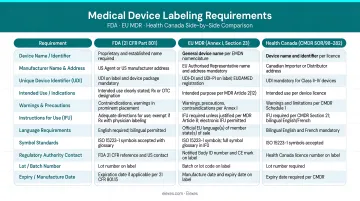
Harmonizing labeling across FDA, EU MDR, ISO 13485, and Health Canada without unnecessary duplication requires strategic planning. Elexes works with manufacturers across these frameworks to build labeling strategies that satisfy each regulatory body's requirements without redundant documentation — reducing both compliance risk and time to market.
Frequently Asked Questions
What are the FDA labeling requirements for medical devices?
All medical devices distributed in the US must comply with 21 CFR Part 801, which requires manufacturer identification (name and address), adequate directions for use, warnings and precautions, UDI (where applicable), and device-specific content such as expiration dates and lot numbers. All labeling materials—not just the physical label—are subject to these requirements.
What is the FDA electronic labeling rule for medical devices?
FDA permits electronic labeling (eIFU) for prescription devices used in healthcare facilities under Section 206 of MDUFMA. This reduces paper-based labeling burdens for facility-based products. OTC devices generally still require paper labeling accessible to consumers.
What are the labeling requirements for investigational devices?
Investigational devices under 21 CFR Part 812 must be labeled with "CAUTION—Investigational Device. Limited by Federal (or United States) law to investigational use," along with sponsor name and address, device name, quantity, and relevant protocols. Investigational devices are exempt from UDI requirements under 21 CFR 801.30(a)(6).
What are the FDA labeling requirements for Class I medical devices?
Class I devices must comply with general labeling requirements under 21 CFR Part 801 (manufacturer info, directions for use, warnings) but may qualify for partial exemptions under Subpart D. Many Class I devices are exempt from UDI under 21 CFR 801.30(a)(2) if they are also exempt from GMP requirements under 21 CFR Part 820.
What are the new FDA label requirements?
The most significant upcoming change is the QMSR transition (effective February 2, 2026), which aligns labeling controls with ISO 13485 while retaining FDA-specific DHR documentation requirements. UDI compliance deadlines are now complete for all device classes (as of 2022), and FDA continues issuing guidance on electronic labeling and SaMD.
What is the difference between FDA QSR and ISO 13485 in terms of labeling controls?
Both FDA QSR (21 CFR 820.120) and ISO 13485:2016 (Sections 7.5.1 and 4.2) require documented labeling controls within the QMS. The key difference: FDA QSR mandates DHR documentation and labeling inspection records with signature and date, while ISO 13485 uses more general verification language under production and service provision controls.


