
Introduction
Medical device complaint handling is a structured, regulatory-mandated process for receiving, evaluating, investigating, and resolving complaints about marketed medical devices to protect patient safety and ensure compliance. Each year, the FDA receives over two million medical device reports of suspected device-associated deaths, serious injuries, and malfunctions—illustrating the sheer volume and criticality of this system.
This guide is designed for QA/RA professionals, medical device manufacturers, startup device companies, IVD makers, and SaMD developers who must establish or optimize their complaint handling operations.
Getting this process right is non-negotiable: it directly affects patient safety, audit outcomes, and continued market authorization. Failures here result in FDA Form 483 observations, warning letters, product recalls, and loss of market access.
What follows covers the full picture: what qualifies as a complaint, how the end-to-end process works, which regulatory frameworks apply, and where companies most often go wrong.
Key Takeaways
- A medical device complaint is any written, electronic, or oral communication alleging a deficiency in a marketed device's identity, quality, safety, reliability, or performance
- The process follows a structured sequence: intake → assessment → investigation → CAPA → closure and trend analysis
- Key regulations include FDA 21 CFR Part 820 §820.198, 21 CFR Part 803 (MDR), ISO 13485:2016 Clause 8.2.2, and EU MDR 2017/745
- Unresolved or mishandled complaints trigger FDA Form 483s, warning letters, recalls, and market withdrawal
- Effective complaint handling drives risk management, post-market surveillance, and continuous product improvement
What Is Medical Device Complaint Handling?
Under FDA 21 CFR 820.3(b), a complaint is "any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a device after it is released for distribution." ISO 13485:2016 Clause 3.4 mirrors this definition — so whether you're targeting FDA clearance or CE marking, the same complaint handling logic applies.
What makes a communication qualify as a complaint versus general feedback? It must:
- Relate to a distributed device (not pre-market)
- Allege a specific deficiency in device characteristics
- Expect a resolution or response from the manufacturer
Not complaints:
- General product inquiries ("How do I use this feature?")
- Internal pre-market observations during validation
- Positive or neutral feedback with no deficiency allegation
- Field incidents that don't allege device fault
Complaint handling sits at the intersection of quality management, post-market surveillance (PMS), vigilance reporting, and CAPA. When poorly managed, the consequences are severe: nonconformances during audits, warning letters, recalls, and revoked market authorization.
A recent example: the FDA issued a warning letter to Mectronic Medicale S.R.L. in June 2025 citing 21 CFR 820.198(a) violations after the firm listed 22 complaints with no evidence they were reviewed or evaluated by a designated unit.
Types of Medical Device Complaints
Manufacturers encounter four primary complaint categories:
Safety Complaints
These involve serious incidents, adverse events, or device malfunctions causing harm. Deaths, serious injuries, or malfunctions likely to cause serious harm if they recurred are MDR-reportable under 21 CFR Part 803.
Quality/Performance Complaints
Manufacturing defects, design flaws, or functional deviations that don't necessarily cause harm but indicate the device fails to meet specifications. These are still investigated under 21 CFR 820.198.
Regulatory Complaints
Issues with labeling non-compliance, missing UDI (Unique Device Identifiers), unapproved marketing claims, or instructions for use deficiencies.
Service/Usability Complaints
User interface issues, inadequate instructions, training gaps, or customer service failures that don't involve device malfunction but allege performance deficiencies.
Correct categorization isn't administrative housekeeping — it drives real consequences. The complaint type determines severity classification, whether an MDR must be filed, and whether immediate containment actions are triggered (product hold, field corrective action, or recall). Misclassification consistently ranks among the most cited causes of FDA 483 observations during complaint handling audits.

The Medical Device Complaint Handling Process: Step by Step
Complaint handling must be embedded within your Quality Management System (QMS) and owned by a formally designated unit—typically QA/RA—with documented SOPs governing each stage. Complaints arrive through multiple channels: phone, email, social media, online forms, customer portals, and in-person reports. All must be captured.
This process is not linear in isolation. Each step remains connected to risk management files, CAPA records, and vigilance reporting obligations throughout.
Step 1: Complaint Intake and Documentation
Intake involves collecting and recording complete, standardized information immediately upon receipt:
- Product identifiers (catalog number, lot/batch, serial number, UDI/DI)
- Complainant details (name, address, phone)
- Date received
- Detailed description of the alleged deficiency
Critical requirement: 21 CFR 820.198(a)(2) mandates that oral complaints must be documented upon receipt. Incomplete intake records routinely cause investigation failures and trigger audit observations.
Step 2: Initial Assessment and Categorization
This step validates whether the report qualifies as a complaint, assigns a severity level (critical, major, moderate, minor), determines complaint type, and evaluates MDR reportability.
Key activities:
- Apply risk-scoring tools (FMEA, severity matrices)
- Determine if the event meets 21 CFR Part 803 MDR thresholds (death, serious injury, or malfunction likely to cause serious harm if it recurred)
- Evaluate EU MDR vigilance requirements (serious incidents, FSCAs (Field Safety Corrective Actions))
- Document escalation decisions immediately
Decisions to escalate or flag for immediate action must be documented. This is where many firms falter—failing to recognize an MDR-reportable event early results in missed deadlines and enforcement action. Once escalation status is confirmed, investigation can begin with the right urgency and scope.
Step 3: Investigation and Root Cause Analysis
Investigation identifies the underlying cause using structured RCA methods:
- 5 Whys – iterative questioning to reach root cause
- Fishbone (Ishikawa) diagrams – categorizing potential causes
- Device history record reviews
- Manufacturing and testing data analysis
- Physical examination of returned devices (if applicable)
Cross-functional teams from QA, RA, engineering, and operations participate. If investigation is deemed unnecessary, 21 CFR 820.198(b) requires documented justification including the name of the individual responsible for the decision. The RCA findings then directly shape the scope and priority of corrective and preventive actions in the next step.
Step 4: CAPA, Resolution, and Communication
CAPA must address both immediate correction and root cause—one fixes the current failure, the other prevents recurrence.
Resolution includes:
- Formally communicating findings to the complainant
- Ensuring privacy compliance
- Filing MDR reports within prescribed timelines (e.g., 30 days for manufacturers under 21 CFR 803.50, 5 days for urgent public health threats under 21 CFR 803.53)
Step 5: Closure, Trend Analysis, and Continuous Improvement
Closure requires verifying CAPA effectiveness before officially closing the record—not just completing paperwork. Aggregated complaint data should be analyzed for patterns across products, markets, or time periods.
Findings feed into:
- Risk management file updates — capturing newly identified failure modes and hazard controls
- Management reviews — providing leadership with trend data to inform resource and quality decisions
- Product design improvements — driving engineering changes that reduce future complaint rates
Done consistently, this analysis turns complaint data into one of the most actionable inputs in your quality system.

Regulatory Requirements for Medical Device Complaint Handling
Complaint handling requirements span multiple jurisdictions — and non-compliance is one of the most commonly cited findings in FDA inspections and MDSAP audits. The core frameworks covered here are FDA 21 CFR Part 820/803/806, ISO 13485:2016, EU MDR/IVDR, and national requirements for the UK, Canada, and Australia.
FDA Requirements
21 CFR Part 820.198 (QMSR):
- Formally designated complaint handling unit
- Documented procedures
- Review of all complaints for MDR reportability
- Investigation or documented justification when investigation is not performed
- Retention of specific investigation records:
- Device ID
- Complaint date
- Complainant contact
- Investigation results
- Corrective actions
- Any reply
21 CFR Part 803 – Medical Device Reporting:
- 30-day reports: Manufacturers must submit reports within 30 days after becoming aware that a device caused or contributed to death, serious injury, or malfunctioned in a way that would be likely to cause serious harm if it recurred (21 CFR 803.50)
- 5-day reports: Required when a reportable event necessitates remedial action to prevent unreasonable risk of substantial harm (21 CFR 803.53)
21 CFR Part 806 – Corrections and Removals:
When complaint investigations reveal systemic issues requiring product corrections or removals, manufacturers must report to FDA within 10 working days (21 CFR 806.10).
FDA QMSR Status:
As of February 2, 2026, FDA finalized the Quality Management System Regulation (QMSR), amending 21 CFR Part 820 to incorporate ISO 13485:2016 by reference and harmonize with international QMS expectations.
ISO 13485:2016 Clause 8.2.2
Minimum requirements include:
- Documented complaint handling procedure
- Evaluation and recording of all complaints
- Investigation or documented justification
- Determination of regulatory reporting need
- CAPA initiation where required
- Maintenance of complaint records
This standard is the foundation for MDSAP audits accepted by FDA, Health Canada, TGA (Australia), ANVISA (Brazil), and PMDA (Japan).
EU MDR and IVDR
Where ISO 13485 sets the global QMS baseline, the EU MDR (Regulation (EU) 2017/745) and EU IVDR (Regulation (EU) 2017/746) impose additional post-market obligations specific to the European market. Both frameworks require complaint handling integrated into the post-market surveillance (PMS) system, with complaints systematically assessed for serious incidents and field safety corrective actions (FSCAs).
Timelines under EU MDR Article 87:
- 2 days: Serious public health threat
- 10 days: Death or unanticipated serious deterioration in health
- 15 days: Other serious incidents
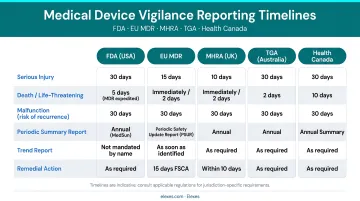
Obligations extend beyond manufacturers to authorized representatives, importers, and distributors.
Other National Frameworks
UK MHRA:
- 2 days: serious public health threat
- 10 days: death or unanticipated serious deterioration
- 15 days: other serious incidents
Source: UK MHRA vigilance reporting requirements
Health Canada:
Mandatory reporting when failure, deterioration in effectiveness, or labeling inadequacy caused or may cause death or serious deterioration in health. Foreign risk notification required within 72 hours (Health Canada industry reporting page).
Australia TGA:
- 48 hours: serious threat to public health
- 10 days: death or serious deterioration
- 30 days: other reportable events
- 120 days: final report
Source: TGA Reporting adverse events for medical devices
Common Mistakes in Complaint Handling (and How to Avoid Them)
Misclassifying Complaints Versus Non-Complaint Feedback
Teams often fail to log borderline reports as complaints or incorrectly dismiss them as field incidents or inquiries. The safest default: log it as a complaint, investigate, and then determine next steps. Misclassification is a top reason for missed MDRs and a frequent trigger for FDA Form 483 observations.
Incomplete Intake Documentation
Vague complaint descriptions, missing product identifiers, or absent complainant details make investigations difficult or impossible. Field technicians and customer service staff receiving complaints must be trained on what information must be captured, down to device-level specifics.
Treating Complaints as Isolated Events Rather Than Data Points
When complaint trend analysis is skipped, systemic quality issues go undetected until they become recalls or regulatory escalations. Complaints should feed into management reviews, risk management file updates, and CAPA systems regularly — not just when a problem becomes obvious.
Poor CAPA Linkage
This systemic gap often shows up most clearly in CAPA handling. Closing a complaint without initiating or verifying meaningful corrective action — especially when root cause has been identified — leaves the underlying risk unresolved.
During inspections, FDA investigators specifically check whether complaint closure is tied to effective corrective action. A complaint record marked "closed" with no linked CAPA, or with a CAPA that was never verified for effectiveness, is a direct path to a 483 observation.
Quick-reference: the four mistakes that most often trigger regulatory findings
- Misclassification — borderline feedback not logged, leading to missed MDR obligations
- Incomplete intake — vague descriptions or missing identifiers that block investigation
- Siloed data — complaints never aggregated for trend analysis or fed into CAPA
- Disconnected CAPA — complaint closed on paper while root cause remains unaddressed

Frequently Asked Questions
What is a medical device complaint?
A medical device complaint is any written, electronic, or oral communication alleging a deficiency in the identity, quality, safety, durability, reliability, effectiveness, or performance of a device after release for distribution, per FDA 21 CFR 820.3(b) and ISO 13485:2016 Clause 3.4.
What is a complaint under 21 CFR 820?
Under 21 CFR Part 820.198, manufacturers must maintain a formally designated complaint-handling unit responsible for receiving, reviewing, and evaluating all complaints. Documented procedures must cover investigation, MDR determination, and record retention — and absence of this system is one of the most common observations in FDA quality system inspections.
What is the complaint handling clause of ISO 13485?
Clause 8.2.2 of ISO 13485:2016 requires documented procedures for complaint evaluation, investigation (or a justified rationale for not investigating), and regulatory reporting assessment. It also mandates CAPA initiation where warranted and defined records retention practices.
What are the types of medical device complaints?
The four main types are safety complaints (device-related adverse events or injuries), quality/performance complaints (defects, failures, or specification deviations), regulatory complaints (labeling or compliance issues), and service/usability complaints (user interface or training-related issues).
What does the medical device reporting regulation require?
FDA 21 CFR Part 803 requires manufacturers to submit MDRs when a device causes or contributes to death, serious injury, or a malfunction likely to cause serious harm if it recurred. Standard reports are due within 30 days; events posing immediate risk require a 5-day expedited report.
What are the 5 stages of complaint handling?
The five core stages are: (1) complaint intake and documentation, (2) initial assessment and categorization including MDR determination, (3) investigation and root cause analysis, (4) corrective action and resolution via CAPA, and (5) closure with trend analysis and continuous improvement review.


