
Introduction
Clinical evaluation deficiencies are among the most frequent reasons notified bodies raise non-conformances during conformity assessment — and most stem from misunderstanding what the guidance actually requires. MEDDEV 2.7/1 Revision 4 is the European Commission's primary guidance document for manufacturers and notified bodies conducting clinical evaluations under Directives 93/42/EEC and 90/385/EEC.
For regulatory affairs professionals, QA/RA teams, and device manufacturers, this matters because clinical evaluation is a continuous process that must evolve throughout a device's lifecycle — not a one-time submission task.
Understanding MEDDEV 2.7/1 Rev 4's expectations remains essential, even as manufacturers transition to EU MDR compliance.
This article explains what MEDDEV 2.7/1 Rev 4 covers, how the clinical evaluation process works, the key requirements it introduced, its continued relevance alongside EU MDR, and common compliance mistakes to avoid.
Key Takeaways
- MEDDEV 2.7/1 Rev 4 (June 2016) covers clinical evaluation across the full device lifecycle, from scoping through CER documentation
- Not legally binding, but treated as accepted best practice and referenced throughout EU MDR MDCG guidance
- Evaluator qualifications, equivalence claims, data validity, and PMS/PMCF linkage all carry higher requirements under Rev 4
- Clinical evaluation follows four stages: scope definition, data identification, data appraisal, and analysis/reporting
- Weak CER documentation and Rev 4 non-compliance are among the most common notified body deficiency triggers
What Is MEDDEV 2.7/1 Revision 4?
MEDDEV 2.7/1 Rev 4 is a guidance document published by the European Commission in June 2016. It provides a common, structured approach for manufacturers and notified bodies to plan, conduct, and document clinical evaluations of medical devices—covering both initial CE marking and ongoing lifecycle updates.
The guidance is designed to ensure devices placed on the EU market have sufficient clinical evidence demonstrating safety, clinical performance, and an acceptable benefit-risk profile relative to the current state of the art.
That said, MEDDEV guidance is not law. As the document itself states: "This guide is not legally binding; only the text of the Directives is authentic in law." In practice, however, it defines accepted best practice and is used by notified bodies to assess clinical evaluation documentation. Non-conformance with MEDDEV 2.7/1 Rev 4 creates a real barrier to CE certification.
Why MEDDEV 2.7/1 Rev 4 Still Matters Under EU MDR
MEDDEV 2.7/1 Rev 4 was written under the old Medical Device Directive (MDD) and AIMDD frameworks. Its core principles for identifying, appraising, and analyzing clinical data still hold up: multiple MDCG guidance documents issued under EU MDR explicitly reference or defer to it.
Key MDCG documents that intersect with MEDDEV 2.7/1 Rev 4:
- MDCG 2020-5 (Clinical evaluation - equivalence): States that "MEDDEV 2.7/1 rev.4 should be used also during the process of demonstrating equivalence under the MDR"
- MDCG 2020-6 (Sufficient clinical evidence for legacy devices): Explicitly lists MEDDEV sections still relevant under MDR in Appendix I
- MDCG 2020-13 (Clinical Evaluation Assessment Report template): References MEDDEV 2.7/1 Rev 4 Section A5 for literature search methods
- MDCG 2019-9 Rev.1 (Summary of Safety and Clinical Performance): Relies on the CER as a primary input, reinforcing the need for a rigorous clinical evaluation methodology
The transition reality: Manufacturers operating under EU MDR must use MDCG documents as primary references, but MEDDEV 2.7/1 Rev 4 remains a valid supplementary resource. This is particularly true for literature search protocols and general CER structure, where detailed MDCG guidance does not yet exist.
EU MDR raises the bar for clinical evidence, particularly for higher-risk and legacy devices. For manufacturers navigating that higher bar, Rev 4's structured methodology for literature search, data appraisal, and clinical analysis remains a practical and regulator-accepted foundation.
How the Clinical Evaluation Process Works Under MEDDEV 2.7/1 Rev 4
MEDDEV 2.7/1 Rev 4 defines a four-stage clinical evaluation process: define scope and objectives, identify and retrieve clinical data, appraise and weight data, and analyze data to produce the CER. This is a continuous lifecycle process, not a one-time activity.
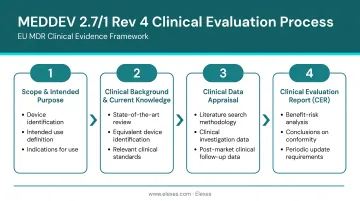
Stage 1: Define Scope and Set Evaluation Objectives
Manufacturers must document:
- Intended purpose, target population, and clinical context
- Indications, contraindications, and intended user profile
- Device description and configuration
- Relevant predicate or equivalent devices
Objectives must be linked to specific safety, performance, and risk-benefit endpoints. Rev 4 clarified that these endpoints must be measurable and tied to the Essential Requirements (now General Safety and Performance Requirements under MDR). Generic objectives like "demonstrate safety" are insufficient: manufacturers must define quantifiable clinical endpoints that allow objective assessment.
Stage 2: Identify and Retrieve Clinical Data
Two primary data streams are required:
Literature searches must be systematic and reproducible, spanning multiple databases such as Embase and PubMed. Key requirements include:
- Documented search strategies using the PICO framework
- Boolean operators with defined inclusion/exclusion criteria
- Full documentation of search terms, databases, and retrieval methodology
- Coverage of Europe-centric databases alongside global sources
Clinical investigations and post-market data — including trials, registries, and post-market surveillance records — must be retrieved and organized with the same level of rigor.
Rev 4 emphasizes that searches must be reproducible: another evaluator should be able to replicate the search strategy and retrieve the same data set.
Stage 3: Appraise and Weight the Data
Each retrieved data source must be evaluated for methodological quality and scientific validity, including:
- Study design and sample size adequacy
- Statistical methodology and potential sources of bias
- Relevance to the specific device and target population
Rev 4 significantly expanded requirements here, requiring manufacturers to justify the weight given to each data type.
Data from equivalent devices is permissible but requires demonstrating equivalence across all three criteria: clinical, technological, and biological.
Stage 4: Analyze Data and Produce the Clinical Evaluation Report (CER)
The CER synthesizes all appraised data into documented conclusions about the device's safety, performance, and risk-benefit profile.
Rev 4 introduced specific CER requirements:
- Author qualifications: Relevant higher education plus minimum five years of experience (or ten years without a degree)
- Declaration of interest for all evaluators
- Defined update frequencies:
- At minimum annually for high-risk or new devices
- Every 2–5 years for lower-risk, well-established devices
Key Requirements Introduced or Clarified in Revision 4
Evaluator Qualifications
Rev 4 introduced explicit criteria for clinical evaluators:
- Relevant higher education degree plus at least five years of related professional experience, OR
- Ten years of experience without a degree
- All evaluators must submit a declaration of interest
Notified bodies now actively scrutinize CER author credentials—this requirement moved from implicit expectation to mandatory documentation.
Equivalence Requirements
What was a footnote in Rev 3 became a detailed appendix in Rev 4. Equivalence must be demonstrated across all three dimensions simultaneously:
- Clinical equivalence: Same clinical indications, contraindications, and intended use
- Biological equivalence: Same materials in contact with the body
- Technological equivalence: Same design, specifications, and working principle

Manufacturers must describe design differences in detail with comparative diagrams.
Beyond diagrams, manufacturers must have documented access to sufficient technical and clinical data for each equivalent device claimed, including competitor devices. Generic statements like "similar to devices on the market" without supporting data access agreements are non-compliant.
Scientific Validity of Data
Rev 4 placed greater emphasis on statistical rigor. Manufacturers must address factors affecting the completeness, objectivity, and weight of data—generic literature citations without methodological appraisal will not satisfy notified body review.
Data sources must be evaluated for:
- Study design appropriateness
- Sample size adequacy
- Risk of bias or conflicts of interest
- Relevance to the specific device and population
Rigorous data appraisal feeds directly into another Rev 4 requirement: keeping the CER current after approval.
PMS and PMCF Linkage
The CER does not end at initial CE marking. Rev 4 requires that post-market surveillance data, including adverse event databases and PMCF activities, feed back into the clinical evaluation and trigger CER updates when new information affects conclusions. This link is reinforced further under EU MDR Article 61.
State of the Art
Rev 4 expanded the requirement to document the state of the art. Manufacturers must characterize not only their own device's safety and performance but also:
- Benchmark devices and alternative treatment options
- Current clinical knowledge and guidelines
- Comparator technologies used in the same clinical context
Without this benchmarking, regulators cannot determine whether a device's benefit-risk profile is acceptable relative to what is already available to patients.
Common Mistakes Manufacturers Make with MEDDEV 2.7/1 Rev 4 Compliance
Relying on a Single Literature Database
Using PubMed alone is the most frequent deficiency cited by notified bodies. Rev 4's Europe-centric requirement means manufacturers must include European databases like Embase. A BSI white paper on clinical evaluation under EU MDR specifically notes that "PubMed-only searches often lack adequate European coverage" and emphasizes that MEDDEV expects multiple databases.
Inadequate database coverage creates a fundamental flaw in the clinical evaluation—it suggests the manufacturer has not conducted a comprehensive search for relevant clinical data.
Using Literature from Non-Equivalent Devices as Clinical Evidence
Data from devices that do not meet all three equivalence criteria can only be used as background context, not as clinical evidence of safety or performance. Two specific failures appear repeatedly in notified body findings:
- Insufficient equivalence documentation: Manufacturers cite literature on "similar" devices without fully documenting equivalence across clinical, biological, and technological dimensions.
- Missing contractual access records: Rev 4 requires documented access to a competitor's technical data to justify equivalence claims. Citing a competitor's instructions for use does not meet this requirement.
Treating the CER as a One-Time Document
Beyond initial submission, the CER must remain a living document. Manufacturers who update it only when prompted by a notified body audit — rather than in response to new PMS data, PMCF findings, or post-market adverse events — are out of conformance and creating patient safety exposure. Rev 4 requires defined update frequencies. Waiting for an audit to trigger a review is not a compliant approach.
Conclusion
MEDDEV 2.7/1 Rev 4 defines a rigorous, systematic approach to clinical evaluation—one that continues to shape how manufacturers build the clinical evidence necessary for EU market access. Whether operating under the MDD legacy framework or transitioning to full EU MDR compliance, manufacturers must understand and apply its core principles.
The guidance rewards structured, reproducible methodology: from scope definition through literature search, data appraisal, and CER documentation. Manufacturers who treat clinical evaluation as a continuous, lifecycle-linked process rather than a pre-market checkbox are better positioned for notified body review and ongoing compliance.
Getting that process right — especially under notified body scrutiny — takes more than a template. Elexes has supported 250+ successful projects across EU MDR, FDA, and global regulatory frameworks, with a 90% audit clearance rate. If you need audit-ready CERs or hands-on clinical evaluation support, their team of regulatory specialists is available on a project or retainer basis.
Frequently Asked Questions
What is MEDDEV 2.7/1 rev 4?
MEDDEV 2.7/1 rev 4 is the European Commission's guidance document for conducting clinical evaluations of medical devices, published in June 2016. It covers clinical data identification, appraisal, and analysis to demonstrate device safety and performance for CE marking under Directives 93/42/EEC and 90/385/EEC.
Is MEDDEV 2.7/1 rev 4 still valid?
Yes, it remains valid and widely used. Even though it was written under the MDD/AIMDD framework, its clinical evaluation methodology is still referenced by MDCG guidance documents under EU MDR—particularly for literature search protocols and CER structure—until more specific MDCG guidance replaces it.
What is the current revision of MEDDEV 2.7/1?
Revision 4, published in June 2016, is the current and most recent version of MEDDEV 2.7/1. No further revision has been officially published, though alignment with EU MDR requirements continues to evolve through MDCG guidance documents.
Are MEDDEV guidance documents still valid?
MEDDEV documents are not legally binding but remain valid as accepted best-practice guidance. MDCG documents are gradually supplementing them under EU MDR, but where no MDCG equivalent exists—as with literature search methodology—MEDDEV guidance continues to apply.
What does the clinical evaluation process involve?
Clinical evaluation involves systematically identifying, appraising, and analyzing clinical data from literature, clinical investigations, or post-market experience to demonstrate a device's safety, performance, and benefit-risk profile. MEDDEV 2.7/1 Rev 4 defines the accepted methodology for this process in the EU.
What is the new Medical Device Regulation (MDR)?
EU MDR (Regulation 2017/745) replaced the old Medical Device Directive, applying stricter requirements for clinical evidence, post-market surveillance, and transparency. While it supersedes the MDD, foundational clinical evaluation guidance from MEDDEV 2.7/1 Rev 4 continues to inform how manufacturers meet MDR's clinical evidence expectations.


