
Introduction
Every medical device carries some degree of risk — and regulators require manufacturers to prove, with documented evidence, that the benefits to patients clearly outweigh those risks before any device reaches the market. A catheter may cause infection; a diagnostic may yield false results; even a Class I device can present hazards if it malfunctions. The question regulators ask isn't "Is this device risk-free?" but rather "Do the clinical benefits justify the residual risks?"
Benefit-risk analysis is a mandatory requirement under ISO 14971:2019, FDA premarket submissions (De Novo, PMA, HDE), and EU MDR 2017/745 across all device classes.
Documentation failures in this area are among the most common reasons submissions face regulatory delays, queries, and rejections. Without a structured, evidence-backed assessment, even a well-designed device can stall at the regulatory gate.
This guide walks through what benefit-risk analysis requires, how to conduct it step by step, and the documentation pitfalls that trip up manufacturers — whether you're filing a first 510(k) or navigating an EU MDR transition.
Key Takeaways
- BRA is mandatory under ISO 14971:2019 Clause 7.4, FDA premarket pathways, and EU MDR/IVDR
- Evidence beats judgment — BRA requires structured, documented evaluation of clinical benefits against residual risks
- Benchmark against state-of-the-art — regulators expect a direct comparison to current treatment alternatives
- Update BRA continuously with post-market surveillance data, complaint trends, and adverse event reports
- Documentation gaps block approvals — inadequate BRA is one of the most cited deficiencies in regulatory submissions
What Is Benefit-Risk Analysis for Medical Devices?
Benefit-risk analysis is the structured process of identifying, quantifying, and comparing the clinical benefits of a medical device against the probability and severity of its associated risks. It's defined in ISO 14971:2019 Clause 7.4 and referenced in EU MDR Article 2(24) as the "analysis of all assessments of benefit and risk of possible relevance for the use of the device for the intended purpose."
Where BRA sits within risk management:
BRA is triggered when residual risks remain after all risk controls have been applied. Under ISO 14971, if the overall residual risk is unacceptable per your risk management plan, you can still justify commercialization if documented clinical benefits outweigh those risks.
Under EU MDR/IVDR, the requirement is broader: BRA applies to every individual risk and the overall residual risk profile, not just unacceptable risks.
BRA vs. risk assessment:
- Risk assessment estimates and evaluates risks (severity × probability)
- Benefit-risk analysis weighs those risks against clinical benefits to reach an acceptability decision
BRA outputs feed directly into both the Risk Management File and the Clinical Evaluation Report (CER), creating traceability between risk, clinical evidence, and regulatory documentation.
US vs. EU terminology:
The FDA historically used "risk-benefit" but revised its guidance in 2019 to place "benefit" first: clinical value drives the analysis, not risk mitigation alone. The FDA's 2019 guidance on benefit-risk determinations for PMA and De Novo submissions formalizes this approach, listing specific factors regulators weigh when evaluating whether a device's benefits justify its risks.
Why Benefit-Risk Analysis Matters in Medical Device Development
Regulatory Compliance Imperative
BRA isn't a "nice to have" — it's a legal requirement enforced globally:
- FDA: Required in De Novo, PMA, and HDE submissions under the 2019 benefit-risk framework
- EU MDR: Required in Technical Documentation (Annex II, Section 5) and Clinical Evaluation (Annex I, Section 1, 3, 8)
- ISO 14971: Recognized by FDA, Health Canada, TGA, and MHLW as the global standard for medical device risk management

A missing or poorly executed BRA can block market access entirely — triggering major deficiency letters, additional information requests, or outright submission rejections.
Patient Safety Dimension
Compliance requirements exist because regulators cannot accept safety on faith. BRA forces manufacturers to confront real-world harms systematically, not assume a device is inherently beneficial. Regulators evaluate:
- Severity of harm (death, serious injury, adverse events)
- Likelihood of occurrence across the indicated population
- Patient tolerance of risk given disease severity
- Medical necessity and availability of alternative treatments
- Duration of exposure and reversibility of harm
These factors must be documented with evidence, not asserted without supporting evidence.
Operational Benefits of a Well-Executed BRA
A thorough BRA delivers measurable downstream benefits:
- Speeds regulatory submissions by giving reviewers a clear, well-supported acceptability rationale
- Reduces post-market recalls by identifying risk control gaps before commercialization
- Supports post-market surveillance by establishing a documented baseline benefit-risk profile against which to measure real-world performance
- Strengthens the CER and Design History File by creating traceability between clinical evidence, risk management, and design decisions
Key Elements of a Benefit-Risk Analysis: Benefits vs. Risks
A BRA has two core inputs: a benefit assessment and a risk assessment, both structured around the device's defined intended use and target patient population.
Benefit Assessment Factors
The FDA's 2019 guidance specifies key benefit factors that must be evaluated:
Type of benefit:
Clinical (treatment efficacy, symptom relief)
Diagnostic (accuracy, sensitivity, specificity)
Quality-of-life improvements
Magnitude of benefit — how significantly the device changes patient outcomes vs. existing standard of care or no treatment
Likelihood of benefit — what proportion of patients in the indicated population are expected to experience it
Duration of effect — whether the benefit is temporary (hours of symptom relief) or sustained (cure, permanent correction)
Patient perspective — how patients value the benefit given their condition severity and available alternatives
Medical necessity — whether comparable alternatives exist; for life-threatening conditions with no effective treatments, even moderate risks may be acceptable
The HeartMate 3 LVAD (PMA P160054) illustrates how magnitude and medical necessity can together justify high residual risks. The device carries serious risks (bleeding, stroke, infection), yet earned approval for advanced heart failure patients because it provides life-sustaining circulatory support with no comparable alternative.
The FDA's SSED documents that probable benefits outweigh probable risks for this population, given disease severity and the absence of therapeutic alternatives.
Risk Assessment Factors
Key risk factors evaluated in a BRA include:
Severity of harm:
Death or serious injury
Non-serious adverse events
Events without reported harm
Likelihood of risk — probability of occurrence and number of patients exposed
Duration of exposure — short-term procedural use vs. chronic implantation
Patient tolerance of risk — patients with life-threatening conditions often accept higher risks than those with mild or self-limiting conditions
Diagnostic consequences — for diagnostics, the clinical impact of false-positive or false-negative results
Contextual factors influencing BRA decisions:
- Uncertainty: Quality and completeness of clinical evidence
- Mitigations: Whether risk controls or labeling adequately reduce residual risk
- Detectability: Whether nonconformity can be identified before use
- Scope of device issue: Isolated cases vs. widespread risk
Two regulatory constraints set hard limits on how risk acceptability can be framed.
EU MDR Annex I, Chapter I, Section 1 requires risks to be acceptable when weighed against benefits "taking into account the generally acknowledged state of the art" — economic or business justifications must not factor into risk acceptability decisions. Separately, the concept of "As Low As Reasonably Practicable" (ALARP), which previously allowed cost considerations in risk reduction decisions, has been removed from ISO 14971:2019. Risk acceptability must now be grounded strictly in clinical benefit, not cost.

How Benefit-Risk Analysis Works – Step by Step
This section translates the regulatory framework into a practical sequence. The most common point of failure: manufacturers treat BRA as a checkbox rather than an ongoing analytical exercise, which reviewers and auditors quickly identify.
Step 1 – Define the Device's Intended Use and Indication
Identify:
- Specific patient population (age, condition, severity)
- Medical condition being addressed
- Clinical context of use (home, hospital, surgical suite)
- Foreseeable misuse scenarios
Why this matters:
The intended use statement anchors all downstream benefit and risk claims. A vague or overly broad intended use is one of the most common root causes of a weak BRA. If your intended use is "cardiovascular monitoring," regulators will expect different evidence than if you specify "post-operative arrhythmia detection in ICU patients."
Step 2 – Identify and Quantify Clinical Benefits
Systematically identify all clinical benefits:
- Treatment efficacy (reduction in symptoms, disease progression)
- Diagnostic accuracy (sensitivity, specificity, predictive values)
- Quality-of-life improvements
- Reduction in procedural time or hospital stay
Support each benefit with objective evidence:
- Clinical trial data
- Peer-reviewed literature
- Registry data
- Real-world evidence
Critical rule:
Benefits without clinical evidence are not acceptable to regulators. Assertions like "may improve patient outcomes" or "expected to reduce complications" without supporting data will trigger deficiency letters.
Step 3 – Identify Hazards and Estimate Risks
Draw on risk analysis outputs from your ISO 14971 process — FMEA, fault tree analysis, preliminary hazard analysis — to complete three tasks:
- Identify all hazardous situations
- Estimate severity and probability of harm for each
- Categorize residual risks after controls
Compare against the state-of-the-art:
How do your device's risks compare to benchmark products or alternative treatments? EU MDR Annex I, Section 1 explicitly requires this comparison. If your new infusion pump has a higher occlusion alarm failure rate than existing pumps, you must document why that risk is acceptable.
Step 4 – Apply Risk Controls and Evaluate Residual Risks
Apply all risk controls in order of priority:
- Inherent safety by design (eliminate the hazard)
- Protective measures in or around the device (guards, alarms, interlocks)
- Information for safety (labeling, instructions for use) — last resort

Evaluate residual risks post-control. BRA should only be applied for risks that remain after controls — not as a substitute for proper risk reduction.
From ISO 14971 Clause 7.1:
The risk control hierarchy is mandatory. If a hazard can be designed out, labeling is not an acceptable risk control.
Step 5 – Conduct the Benefit-Risk Comparison and Reach a Conclusion
Compare benefits and residual risks across three dimensions:
- Magnitude — severity of benefits versus seriousness of the risks involved
- Frequency/Probability — the proportion of patients who benefit compared to those who experience harm
- Duration — whether benefits and harms are temporary or long-lasting
The conclusion must be explicit:
State whether the benefit-risk profile is acceptable and document the rationale. Avoid vague conclusions like "benefits are believed to outweigh risks." Instead: "For patients with Stage IV heart failure and no surgical options, the 15% reduction in 1-year mortality outweighs the 8% risk of infection, given the absence of comparable alternatives."
Step 6 – Document in the Risk Management File and Update Post-Market
Record the BRA outcome in the Risk Management File and cross-reference it with the Clinical Evaluation Report.
BRA is a living document:
It must be updated whenever:
- Post-market surveillance data reveals new risks or changes in risk frequency
- Complaint trends indicate previously unidentified hazards
- Adverse event reports suggest benefit-risk profile shifts
- New clinical evidence (literature, registries) changes the state-of-the-art
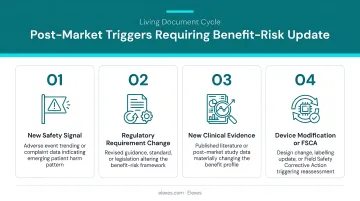
Manufacturers who neglect this step after commercialization are a common audit finding. ISO 14971 Clause 10 requires production and post-production information review. Separately, EU MDR Annex I, Section 3(e) mandates evaluating how post-market surveillance data affects the overall benefit-risk ratio.
Common Mistakes in Benefit-Risk Analysis Documentation
Mistake 1: Conducting BRA in Isolation from Clinical Evaluation
Regulators expect traceability between the Risk Management File and the CER. Each residual risk should link back to clinical evidence demonstrating that the benefit outweighs it. If your RMF states "risk of device migration is acceptable because clinical benefit outweighs it," the CER must contain the clinical data supporting that claim.
MDCG 2020-13 Clinical Evaluation Assessment Report template explicitly requires documenting alignment between clinical evidence, GSPRs, and benefit-risk conclusions.
Mistake 2: Relying Solely on Qualitative Reasoning Without Quantitative Parameters
Beyond traceability, regulators — particularly under EU MDR — expect measurable outcomes to support benefit-risk conclusions:
- Complication rates (%)
- Success rates (%)
- Comparative device performance data from state-of-the-art benchmarks
From MEDDEV 2.7/1 Rev. 4:
Clinical evaluation must include quantitative parameters and explicit state-of-the-art benchmarking. Statements like "device is expected to perform similarly to predicate devices" without supporting data do not meet the standard.
Mistake 3: Treating BRA as a Static, One-Time Document
Even well-supported analyses can fail post-market if the BRA is never updated. Completing BRA during design and leaving it unchanged is a common finding in regulatory audits and notified body assessments. Post-market surveillance data, adverse event trends, and updates to the state-of-the-art must all feed back into an updated BRA throughout the device's commercial life.
Real-world conditions that invalidate an existing BRA include:
- Post-market complication rates exceeding clinical trial estimates
- A competitor launching a device with a materially better safety profile
- New clinical evidence that shifts the state-of-the-art benchmark
In any of these scenarios, regulators expect re-evaluation of risk acceptability and corresponding updates to both the RMF and CER.
How Elexes Can Help
Elexes is a regulatory consulting partner for medical device manufacturers who need expert support building, reviewing, or remediating benefit-risk analysis documentation. With 50+ years of collective experience and deep ISO 14971 expertise, Elexes helps manufacturers structure BRA that withstands regulatory scrutiny across the full product lifecycle.
The firm's track record spans 250+ successful projects across FDA, EU MDR, Health Canada, TGA, and other global regulatory bodies — covering everything from first submission through post-market lifecycle management.
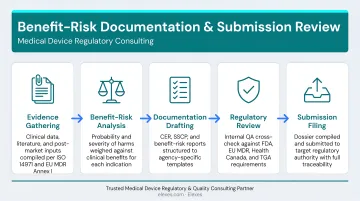
Services directly supporting BRA:
- Risk Management File development and gap assessment: Builds or remediates your RMF to meet ISO 14971:2019 and regional regulatory requirements
- Clinical Evaluation support (CER/CEP): Integrates benefit-risk analysis with clinical evidence and state-of-the-art benchmarking
- Regulatory submission preparation: Structures compliant BRA sections for PMA, De Novo, 510(k), and EU MDR Technical Documentation
- Post-market surveillance integration: Connects complaint handling and PMS outputs directly into risk management file updates
All services are delivered with full confidentiality on a project-based, retainer, or embedded-consultant model depending on your development stage and regulatory pathway.
For medical device companies (especially startups and companies entering new markets), engaging Elexes early in the development cycle helps avoid the BRA documentation gaps most likely to delay approvals.
Contact Elexes to get a gap assessment of your benefit-risk documentation and confirm it's submission-ready.
Frequently Asked Questions
What should a risk-benefit analysis include?
A benefit-risk analysis should include:
- Clearly defined intended use and patient population
- Evidence-based benefit assessment (type, magnitude, likelihood, duration)
- Risk assessment covering severity, probability, and patient tolerance
- Comparison of residual risks against benefits
- Documented conclusion cross-referenced with the Clinical Evaluation Report and Risk Management File
What are the four steps of a risk-benefit analysis?
The four key steps are:
- Identify the intended use and patient population
- Identify and quantify clinical benefits using clinical evidence
- Identify and assess residual risks after applying risk controls
- Compare benefits against risks and document the acceptability conclusion
When is a benefit-risk analysis required for medical devices?
Under ISO 14971, BRA is required when residual risks remain unacceptable after controls. Under EU MDR/IVDR, it is required for every individual risk and all residual risks regardless of risk level. Under FDA, it is required for De Novo, PMA, and HDE submissions.
How does ISO 14971 relate to benefit-risk analysis?
ISO 14971:2019 Clause 7.4 defines the benefit-risk analysis requirement within the risk management process, specifying that if residual risks are unacceptable, manufacturers can still justify commercialization if documented clinical benefits outweigh the risks, and that financial considerations must not factor into this determination.
What is the difference between benefit-risk analysis under FDA and EU MDR?
FDA requires BRA for high-risk submissions (PMA, De Novo, HDE) and evaluates it case-by-case using factors like medical necessity and patient tolerance. EU MDR requires BRA for all risks across all device classes, mandates quantitative parameters, and embeds the analysis in both the Risk Management File and Clinical Evaluation Report.
Does benefit-risk analysis need to be updated after a device is on the market?
Yes, BRA must be treated as a living document. Post-market surveillance data, adverse event reports, complaint trends, CAPA outcomes, and changes to the state-of-the-art must all be evaluated for their impact on the benefit-risk profile, and the Risk Management File and CER updated accordingly.


