
Introduction
Manufacturers of legacy devices—those certified under the Medical Device Directive (MDD) or Active Implantable Medical Device Directive (AIMDD) and still on the EU market under transitional provisions—face a critical compliance question with every device modification: does this change void the existing certificate?
Industry data from 2022 showed that more than 85% of legacy products had not yet received MDR certificates — a gap that confirms how broadly this issue affects manufacturers today.
MDCG 2020-3 exists to answer the significant change question directly. Misreading a change as non-significant can expose a manufacturer to serious consequences, including immediate loss of market authorization. This guide walks through how the guidance works and what your team needs to assess it correctly.
Key Takeaways
- MDCG 2020-3 defines "significant change" in design or intended purpose for legacy devices under Article 120 MDR
- A significant change invalidates your MDD/AIMDD certificate and triggers full MDR certification
- The guidance applies a two-part test: the change must concern design or intended purpose AND be significant
- Five sub-charts (A–E) cover intended purpose, design, software, materials, and sterilization changes
- Manufacturers must document change assessments and bear the burden of proof for competent authority review
What Is MDCG 2020-3 and Why Does It Matter?
MDCG 2020-3 is guidance published by the Medical Device Coordination Group (MDCG) to clarify what constitutes a "significant change in design or intended purpose" for legacy devices under Article 120(3) of EU MDR 2017/745. The MDCG is composed of representatives from all EU Member States and is chaired by the European Commission.
Who Does This Guidance Apply To?
This guidance applies to manufacturers of legacy devices: devices placed on the EU market after 26 May 2021 under a still-valid MDD or AIMDD certificate, taking advantage of the extended transitional deadlines established by Regulation (EU) 2023/607:
- 31 December 2027 for Class III devices and Class IIb implantables (except sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips, and connectors)
- 31 December 2028 for other Class IIb devices, Class IIa, and sterile/measuring Class I devices
Legal Weight
The guidance is not legally binding in itself. Only the Court of Justice of the EU can provide binding interpretations of Union law. That said, it represents agreed regulatory best practice endorsed by all EU Member States, and notified bodies and competent authorities apply it during surveillance and audits.
Article 120 of EU MDR: The Transitional Provision Explained
Article 120(3) MDR (as amended by Regulation (EU) 2023/607) permits legacy devices to remain on the market past the MDR's date of application, provided two key conditions are met simultaneously:
- The device continues to comply with the applicable Directive (AIMDD or MDD)
- There are no significant changes in the design or intended purpose of the device
The Critical Certificate Constraint
Section 3 of MDCG 2020-3 establishes a clear restriction: no new or modified/amended MDD or AIMDD certificates may be issued during the transitional period. This means manufacturers cannot simply update or reissue a directive certificate to accommodate a significant change. The only pathway forward is full MDR certification.
Role of the Notified Body
Within this constraint framework, notified bodies verify changes as part of surveillance activities or following a manufacturer's prior approval submission. When a change is confirmed as non-significant, the notified body may issue a written confirmation that the existing certificate remains valid. This is not a supplemental certificate — it is a verification statement only, not a new or amended directive certification.
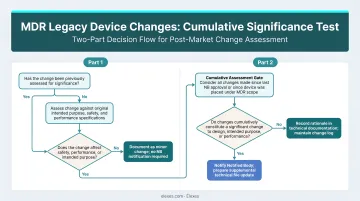
The Two-Part Significance Test Under MDCG 2020-3
MDCG 2020-3 establishes a two-part cumulative test:
- There must be a change that concerns the design or intended purpose
- That change must be significant
Changes that do not touch design or intended purpose at all are entirely out of scope of Article 120(3) MDR.
What Falls Outside the Scope
Administrative and organizational changes do not affect design or intended purpose and are therefore out of scope, including:
- Manufacturer name, address, or legal form changes (same legal entity)
- Authorized representative changes
- Relocation or addition of manufacturing sites under unchanged certification conditions
- Supplier changes within unchanged specifications
Note: These changes may still require notification to the notified body under existing AIMDD/MDD contractual arrangements.
Explicit Carve-Outs
Changes for regulatory compliance: Changes made solely to comply with other Union legislation (such as REACH regulation substance substitutions) are out of scope, provided the risk/benefit ratio is not negatively affected.
Corrective action exception: Design or intended purpose changes related to corrective actions assessed and accepted by the relevant competent authority are treated as non-significant—including field safety corrective actions (FSCAs).
Burden of Proof
Manufacturers are responsible for documenting their assessment and providing evidence that a change is non-significant. Manufacturers must make this justification available to competent authorities on request. In cases of doubt, consult your notified body.
Category-by-Category Breakdown: What Counts as Significant?
MDCG 2020-3 uses a flowchart methodology: a main chart plus five sub-charts (A through E), each addressing a specific category of change. A change is non-significant only if every applicable sub-chart resolves to "non-significant." One "significant" answer in any sub-chart determines the overall outcome.
The five sub-charts cover the following categories:
| Chart | Category | Key Focus |
|---|---|---|
| A | Intended Purpose | Indications, populations, anatomical sites |
| B | Design & Performance | Operating principles, energy sources, specifications |
| C | Software | Algorithms, architecture, UI, interoperability |
| D | Substances & Materials | Contact materials, biologics, medicinal substances |
| E | Sterilization | Method, packaging, shelf life, storage conditions |
Changes in Intended Purpose (Chart A)
Intended purpose changes are among the most closely scrutinized — expanding scope in almost any direction triggers a significant classification.
Non-significant changes:
- Any restriction or limitation: narrowing indications, deleting clinical applications or anatomical sites, restricting target patient populations
- These reduce rather than expand the device's scope
Significant changes:
- Extensions or alterations in any direction: adding new indications or clinical conditions
- Targeting new user or patient populations (for example, professional use to lay users)
- Changing anatomical site, delivery pathway, or deployment method
- Labelling changes linked to intended use (contraindications, warnings)
Changes in Design and Performance Specifications (Chart B)
Chart B draws a clear line between cosmetic or compliance-driven modifications and changes that touch the device's core operating logic.
Non-significant changes:
- Modifications that do not alter built-in control mechanism, operating principle, energy source, or alarm systems
- Changes that do not adversely affect safety, performance, usability, or risk/benefit ratio
- Examples: new variants within certified dimensional ranges (for example, intermediate stent lengths), ergonomic grip modifications, minor PCB re-layout due to component obsolescence, packaging changes for non-sterile devices
Triggers for notified body review:
- Modification to built-in control mechanism, operating principle, energy source, or alarm systems
- Switching from analog to digital control, changing measurement wavelength, adding/removing alarm systems
- Expanding specification limits on critical components
- Any change adversely affecting safety or performance that negatively impacts the risk/benefit ratio (for example, wire diameter reduction outside certified specs, adding electrodes to an implantable lead)

Software Changes (Chart C)
Software is evaluated on whether a change affects operating principles, clinical output, or device control — not just on whether code was modified.
Non-significant changes:
- Bug fixes restoring original specifications
- Performance improvements reducing latency
- Cybersecurity updates and security patches
- Operating system updates not requiring device software modification (for example, Windows security updates)
- UI appearance changes not affecting usability or adding new functions
- New non-medical features: improved reporting format, additional language support, barcode reader functionality
Significant changes:
- Modifications to architecture, database structure, or core algorithms impacting operating principles or device control
- Replacement of required user input with a closed-loop algorithm
- Major OS changes requiring device software modification (for example, Windows 10 to Windows 11 if modification required)
- New user interfaces changing how medical data is read or interpreted (for example, keyboard to touchscreen for a critical function)
- New medical features or functionalities that may alter diagnosis or therapy
- New channels of interoperability
Changes in Substances or Materials (Chart D)
Chart D focuses on biocompatibility risks, with the 30-day patient-contact threshold serving as a key classification trigger for implant and long-term contact materials.
Non-significant changes:
- Supplier changes for existing materials within unchanged specifications
- Substitutions required for regulatory compliance (for example, REACH)
- Changes to processing aids not present on the final cleaned device
- Material grade updates without specification changes
Significant changes:
- Changes to materials intended for direct or indirect contact with patient tissue or fluid for more than 30 days (implant materials)
- Addition or substitution of materials of human or animal origin
- Changes to medicinal substances or excipient carriers
- Any material change increasing toxicological or biological risk, or adversely affecting safety or performance
Changes Related to Sterilization (Chart E)
Non-significant changes:
- Adjustments to sterilization cycle parameters within approved QMS protocols
- New sterilization vendor or chamber without changing method
- Change from single to double sterile packaging
- Shelf-life extensions validated against notified body-approved protocols
Significant changes:
- Changing terminal sterilization method (for example, ETO to Gamma)
- Switching from biological indicator to parametric release
- Changing device labelling from non-sterile to sterile
- Packaging design changes adversely affecting sterility or seal integrity
- Shelf-life extensions not validated against approved protocols
- Storage or transportation condition changes that could adversely affect sterility or stability

What Happens When a Change Is Deemed Significant
If a change is classified as significant, the manufacturer can no longer place the device on the EU market under the existing MDD or AIMDD certificate. The device must be brought into full conformity with EU MDR 2017/745, which means selecting the appropriate MDR conformity assessment route for the device's classification (for example, Annex IX, X, or XI) and obtaining an EU MDR certificate before continuing to market.
Two Practical Options
Manufacturers facing this situation have two paths:
- Pause and complete MDR certification before implementing the change
- Forgo the change if MDR certification cannot be achieved in time
Either path requires a clear regulatory plan: notified body engagement, updated technical documentation, and in most cases a clinical evaluation update. Working with experienced EU MDR regulatory consultants early in the process reduces delays and limits the risk of non-compliance during the transition.
Downstream Impact
Manufacturers should evaluate the downstream impact:
- UDI updates and EUDAMED registration
- Post-market surveillance plan revisions
- Clinical evaluation report updates
- Quality management system documentation changes
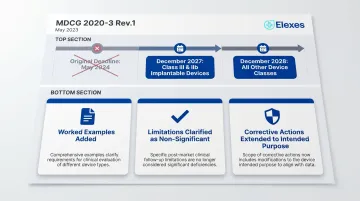
Key Updates in MDCG 2020-3 Rev.1 (May 2023)
The Rev.1 revision, published in May 2023, was driven by two factors: alignment with Regulation (EU) 2023/607, which extended the transitional deadlines, and harmonization with MDCG 2022-2 guidance. With deadlines pushed to December 2027 and December 2028 (from the original May 2024), manufacturers now have more time — but also a longer window during which legacy certificates must remain compliant, making accurate significant change assessments more consequential than ever.
Substantive Improvements in Rev.1
- Worked examples added throughout each category (Charts A–E) make flowchart decisions more practical and reduce ambiguity in borderline cases
- General principles now explicitly acknowledge that changes in intended purpose resulting in a limitation or restriction are non-significant — a previously unclear area
- Corrective action changes accepted by competent authorities now explicitly cover intended purpose changes, not just design changes
Parallel IVDR Guidance
MDCG 2022-6, published in May 2022, applies the same significant change principles to IVDs under Article 110(3) of the IVDR. It provides IVD-specific examples (such as changes to PCR primers, antibodies, or assay methods). Manufacturers of IVDs should consult both documents together, as MDCG 2022-6 builds directly on the framework established in MDCG 2020-3.
Frequently Asked Questions
What is MDCG 2020-3?
MDCG 2020-3 is a guidance document published by the Medical Device Coordination Group that clarifies what constitutes a "significant change in design or intended purpose" for legacy devices under Article 120 of EU MDR, helping manufacturers determine whether a change to an MDD/AIMDD-certified device will invalidate that certificate.
What is a significant change under MDR?
Under MDCG 2020-3, a significant change requires two cumulative conditions: the change must concern the design or intended purpose, and it must be significant in nature. Examples include new indications, new user populations, core operating mechanism changes, major software architecture changes, or implant material changes.
Is the MDCG legally binding?
MDCG guidance documents are not legally binding—only the Court of Justice of the EU can provide binding interpretations of Union law. However, they represent agreed best practice endorsed by all EU Member States and are actively used by notified bodies and competent authorities when assessing compliance.
What is Article 120 of the EU MDR?
Article 120 of EU MDR 2017/745 (as amended by Regulation (EU) 2023/607) contains the transitional provisions for legacy devices. Devices with valid MDD or AIMDD certificates may remain on the EU market until December 2027 or December 2028 (depending on classification), provided no significant changes are made to design or intended purpose.
What is the MDCG for medical devices?
The MDCG (Medical Device Coordination Group) is a body established under Article 103 of EU MDR, composed of representatives from all EU Member States and chaired by the European Commission, tasked with providing guidance and ensuring consistent application of EU MDR and IVDR across member states.


