
Introduction
Pursuing ISO 13485 certification without conducting a thorough gap analysis is one of the most common—and costly—mistakes medical device companies make. Without this diagnostic step, organizations routinely face failed audits, delayed market entry, and expensive rework that can push timelines back by months. According to the ISO Survey, over 27,000 ISO 13485 certificates were issued globally in 2021, yet a significant share of those initial certification attempts hit avoidable obstacles — documentation gaps, undocumented processes, and missing risk controls that a pre-audit review would have caught.
This guide covers what an ISO 13485 gap analysis is, why it's non-negotiable for QMS compliance, how to conduct one clause by clause, and how to transform your findings into a structured remediation plan that keeps your certification on track.
Key Takeaways
- Gap analysis identifies what your QMS is missing before a formal audit catches it
- Applies to both first-time certification candidates and teams maintaining ongoing compliance
- Covers scoping, document review, clause-by-clause assessment, and corrective action planning
- Clauses 7 and 8 generate the most non-conformances in assessments
- Done well, it cuts audit failure risk and shortens the path to certification
What Is ISO 13485 Gap Analysis?
ISO 13485 gap analysis is a structured evaluation that compares your current Quality Management System against A gap analysis compares your current Quality Management System against the requirements of ISO 13485:2016 to identify areas of non-conformance, partial conformance, or missing documentation and processes. It establishes a compliance baseline before you engage a certification body for formal audit.
What it is NOT:
- Separate from the certification audit itself
- Not a substitute for internal audit (required under Clause 8.2.4)
- A point-in-time exercise, not an ongoing compliance mechanism
Think of it as a diagnostic that tells you where you stand before the formal process begins. It applies in two distinct situations:
- First-time certification: Identifies the distance between current practices and ISO 13485 requirements, so you can scope implementation work accurately
- Recertification or surveillance preparation: Used by certified organizations ahead of audits, after QMS changes, or when aligning with regulatory updates — such as the FDA's 2024 QMSR final rule
Why ISO 13485 Gap Analysis Matters for Medical Device Companies
Gap analysis directly determines audit readiness. Companies that skip or poorly execute this step face significant business consequences:
- Delayed CE marking or market authorization when non-conformances surface during certification
- Regulatory findings during formal audits that trigger corrective action requirements
- Product recalls stemming from undetected QMS failures
- Increased time-to-market due to re-audit cycles
Done well, gap analysis turns those risks into a structured remediation roadmap before auditors ever walk through the door:
- Identifies compliance risks before they become formal audit findings
- Provides a clear baseline for QMS improvement planning
- Prioritizes remediation based on gap severity and patient safety risk
- Reduces certification cost by minimizing re-audit cycles
- Demonstrates due diligence to regulators and notified bodies
How to Conduct an ISO 13485 Gap Analysis – Step by Step
Gap analysis in medical devices requires precision beyond general ISO standards because regulatory consequences are directly tied to patient safety. Each step should be treated as a quality activity, not a compliance checkbox.
Step 1 – Define Scope and Objectives
Identify which parts of your organization, processes, and product lines fall within the ISO 13485 QMS scope. Scope definition directly affects which clauses apply and which may be excluded.
Example: Clause 7.3 design controls can be excluded if design is outsourced, provided this exclusion is documented per Clause 4.2.2 and applicable regulatory requirements permit it. However, even when outsourced, you remain responsible for controlling that process under Clauses 4.1.5 and 7.4.
Step 2 – Gather Existing QMS Documentation
Collect all current quality documentation:
- Quality manual
- Procedures and work instructions
- Process maps and flow charts
- Quality records and device files
- Supplier agreements and approved supplier lists
Many companies have processes in practice but lack the documented evidence ISO 13485 requires. Document completeness is often the first major finding.
Step 3 – Map Current Practices Against ISO 13485 Clauses
Conduct a clause-by-clause assessment across all applicable sections (Clauses 4 through 8). ISO 13485:2016 is structured into five main sections:
- Clause 4: Quality management system (general requirements, documentation, medical device file, document/records control)
- Clause 5: Management responsibility (policy, planning, authority, management review)
- Clause 6: Resource management (personnel, infrastructure, work environment)
- Clause 7: Product realization (planning, customer processes, design, purchasing, production, equipment control)
- Clause 8: Measurement, analysis, improvement (feedback, complaints, regulatory reporting, internal audit, CAPA)
For each requirement, use a structured checklist or matrix to record one of three conformance statuses:
- Fully conforming — evidence exists and meets the requirement
- Partially conforming — some elements present but gaps remain
- Non-conforming — requirement not met or no evidence exists
Step 4 – Interview Key Personnel and Observe Processes
Actual practice often differs from documented procedures — direct observation surfaces those discrepancies before an auditor does. Speak with employees in:
- Production and manufacturing
- Design and development
- Procurement and supplier management
- Post-market surveillance and complaint handling
- Quality assurance and regulatory affairs
Step 5 – Document and Categorize Findings
Record all identified gaps with:
- Severity level: critical non-conformance, minor non-conformance, or opportunity for improvement
- ISO 13485 clause reference with the specific requirement cited
- Evidence reviewed: documents, interviews, or direct observations
- Current state vs. required state: what exists and what's needed
Step 6 – Prioritize and Report
Organize findings into a gap analysis report that ranks remediation priorities based on:
- Patient safety risk
- Audit criticality (likelihood of being flagged as major non-conformance)
- Implementation effort and resources required
This output becomes the foundation for your corrective action and implementation plan.
Key ISO 13485 Clauses Commonly Flagged in Gap Analyses
While every clause must be assessed, certain sections consistently generate the highest number of non-conformances—especially for companies new to the standard or transitioning from ISO 9001.
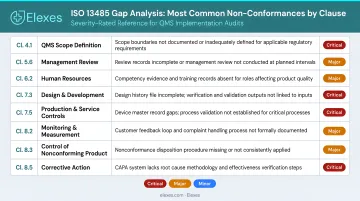
Clause 4.1 (General QMS Requirements) and Clause 4.2 (Documentation Requirements)
Clause 4.2 specifically requires:
- Quality manual
- Documented procedures (six mandatory procedures specified throughout the standard)
- Medical device file
- Control of documents and records
Missing or incomplete documentation is the most frequent failure here. Without it, you cannot demonstrate compliance in any other clause — making this the most foundational gap to close first.
Clause 7.3 (Design and Development)
Clause 7.3 covers the full design lifecycle, and it's where traceability breakdowns are most costly:
- Design planning, inputs, outputs
- Design review, verification, validation
- Design transfer
- Design and development file (Clause 7.3.10) — added in 2016
The most common failure is broken traceability between design inputs and outputs. Companies frequently lack a complete design history file with records that demonstrate conformity at each stage.
Clause 7.4 (Purchasing and Supplier Control)
ISO 13485:2016 significantly strengthened supplier management requirements, including:
- Risk-based supplier evaluation and re-evaluation
- Written quality agreements with suppliers
- Supplier change notification agreements
- Verification of purchased product proportionate to risk
In practice, supplier evaluation criteria are rarely tied to device risk, and records of ongoing supplier monitoring are either incomplete or absent. Both issues surface regularly in audits.
Supplier gaps often have downstream effects on complaint handling — which is where the next clause comes in.
Clause 8.2 (Feedback, Complaint Handling, and Reporting)
The 2016 revision added specific requirements for:
- Complaint handling procedure (8.2.2)
- Reporting to regulatory authorities (8.2.3)
- Feedback into risk management
Many organizations handle complaints informally — no documented process for capture, evaluation, investigation, or escalation. Regulatory reporting procedures (8.2.3) are also missing more often than not, a gap auditors routinely flag.
Clause 8.5 (Improvement – CAPA)
CAPA systems are frequently found incomplete during gap analysis. The standard requires:
- Documented root cause analysis
- Implementation of corrective actions without undue delay
- Verification of effectiveness
- Assessment of whether actions adversely affect other processes or products
The core failure: corrective actions get implemented, but without documented root cause analysis, effectiveness verification, or any assessment of impact on other devices or processes. The action gets closed — the problem often doesn't.
Turning ISO 13485 Gap Analysis Findings into an Action Plan
A gap analysis report only drives results when it becomes a concrete remediation plan. For each identified gap, your action plan should document:
- Who owns it: Name the specific person accountable — not a team or department
- When it closes: Set realistic deadlines that account for document approval cycles and required training time
- What's needed: List procedure writing, training, software, or infrastructure requirements up front
- How closure is confirmed: Define the evidence required — such as updated SOPs, training records, or completed forms
Prioritization Framework
Address first — Critical non-conformances:
- Patient safety-related gaps
- Legally mandated procedures (complaint handling, CAPA, document control)
- Major non-conformances that would delay certification
Address second — Minor non-conformances:
- Documentation gaps that don't directly impact patient safety
- Process improvements that strengthen compliance
Address later — Opportunities for improvement:
- Enhancements beyond minimum requirements
- Efficiency improvements
Maintain Evidence as Gaps Are Closed
Every remediated gap must be supported by documented evidence:
- Updated SOPs and work instructions
- Training records showing personnel competence
- Completed forms demonstrating process implementation
- Records proving effectiveness of corrective actions
This evidence trail is what you'll present to an auditor. Many companies complete remediation work but fail to maintain the documentation that proves closure.
Integrate Periodic Gap Reviews
Gap analysis works best as an ongoing discipline, not a pre-audit scramble. Integrate periodic internal gap reviews into your QMS management review cycle (Clause 5.6) to maintain continuous audit readiness — and avoid the compressed timelines that come with leaving it until certification is imminent.
How Elexes Supports ISO 13485 Gap Analysis for Medical Device Companies
Elexes is a regulatory affairs and QMS consulting firm with 50+ years of collective experience conducting ISO 13485 gap analyses for medical device companies—from startups building QMS from scratch to established manufacturers preparing for recertification or market expansion into new regions (FDA, EU MDR, Health Canada, MDSAP).
What Elexes brings to gap analysis engagements:
- Clause-by-clause assessment against ISO 13485:2016
- Risk-based prioritization of findings tied to patient safety and regulatory criticality
- Development of structured remediation roadmap with assigned ownership and milestone timelines
- Hands-on support closing documentation and process gaps
- Proven track record across 250+ completed projects with a 90% audit clearance rate

Regardless of where you are in the certification cycle — first-time certification, post-QMS change, or expanding into a new regulatory market — earlier engagement with an experienced consultant cuts certification timelines and reduces the risk of costly non-conformances during formal audits.
Ready to assess your ISO 13485 compliance baseline? Connect with Elexes for a gap assessment consultation.
Frequently Asked Questions
What is ISO gap analysis?
An ISO gap analysis is a systematic comparison of your organization's current practices and documentation against the requirements of a specific ISO standard. In the context of ISO 13485, it identifies where your QMS falls short of what the standard requires before a formal certification audit.
What are the 4 steps in a gap analysis?
The four core steps are:
- Define target requirements based on the ISO standard or quality goals
- Assess current processes and documentation against those requirements
- Identify and quantify gaps between the current state and the required state
- Plan remediation actions to close each identified gap
What is a gap analysis checklist?
A gap analysis checklist is a structured document that lists all ISO 13485 clause requirements, allowing assessors to evaluate and record compliance status for each item. It serves as both the assessment tool and an audit trail for the review.
What does clause 4.2 of ISO 13485 provide details of?
Clause 4.2 covers documentation requirements for the QMS: the quality manual, required documented procedures, the medical device file, control of documents, and control of records. These elements form the foundational documentation framework that all other clauses build upon.
How long does an ISO 13485 gap analysis take?
The timeline depends on organization size, QMS maturity, and scope. A focused assessment for a small medical device company may take one to two weeks, while a complex multi-site organization could require four to eight weeks. Organizations with more complete existing documentation tend to move through the process faster.
What is the difference between an ISO 13485 gap analysis and an internal audit?
A gap analysis is a diagnostic tool used to identify what's missing or non-conforming before implementing or improving a QMS. An internal audit verifies whether an already-established QMS is functioning as intended.


