

Medical Device Risk Analysis: ISO 14971 Compliance Guide
Before we discuss how often the risk review must be conducted, let’s understand what risk is, why risk analysis is important, and look at the different tools involved.
Elexes has helped 100+ companies build FDA/ISO 14971-compliant risk files for software, hardware, and combination devices. If you’re a QA/RA head or product engineer, this guide will help you build risk files that pass regulatory audits.
What is meant by risk and what is the importance of risk analysis?
In a medical device company, “risk” refers to the potential for harm or adverse outcomes associated with the use of a medical device.
The most common risks associated with medical devices are risks to patient safety, regulatory compliance, financial stability, and reputation. Each one of these risks can cause potential harm and, therefore, lead to legal issues for a company.
Considering this, we all agree that performing regular risk review is critical for every medical device company. By conducting proper risk analysis and periodic risk reviews, companies can detect issues and resolve them before they become major problems.
What are the tools used for risk analysis?
Several tools are available today that can help in medical device risk analysis. Some of these tools are –
⦿ Primary Hazards Analysis (PHA)
⦿ Event Tree Analysis
⦿ Fault Tree Analysis
⦿ Failure Mode and Effects Analysis (FMEA)
To understand more about these medical device risk analysis tools, you must go through this post drafted by our CEO Parul Chansoria –
Risk Management Methods for Medical Devices
In this post, you will find all the details on each of the risk analysis tools, including their pros and cons. We highly recommend you check this post to learn more about these medical device risk analysis and management tools.
While there exist several tools as detailed above; in this blog, we will explain everything about risk analysis and review by utilizing one of these methods: FMEA.
So let’s dive right in…..
What is FMEA?
Failure Mode and Effects Analysis (FMEA) is a systematic method for analyzing potential failure modes of a product or a process by assessing their effects on system performance. This process involves.
⦿ Identifying failure modes
⦿ Determining their causes and effects
⦿ Assigning severity, occurrence, and detection ratings
⦿ Calculating a Risk Priority Number (RPN) to prioritise risks for mitigation
Frequency of conducting Risk Review:
Risk reviews should be conducted multiple times a year, as needed. The frequency depends on how often changes, feedback, or updates occur within the company. The goal is to keep the risk analysis created using the FMEA tool a dynamic document that accurately reflects current product and process risks.

NOTE: Risk analysis and review are important throughout the product life cycle, and they should be done thoroughly every time.
What changes trigger Risk Review?
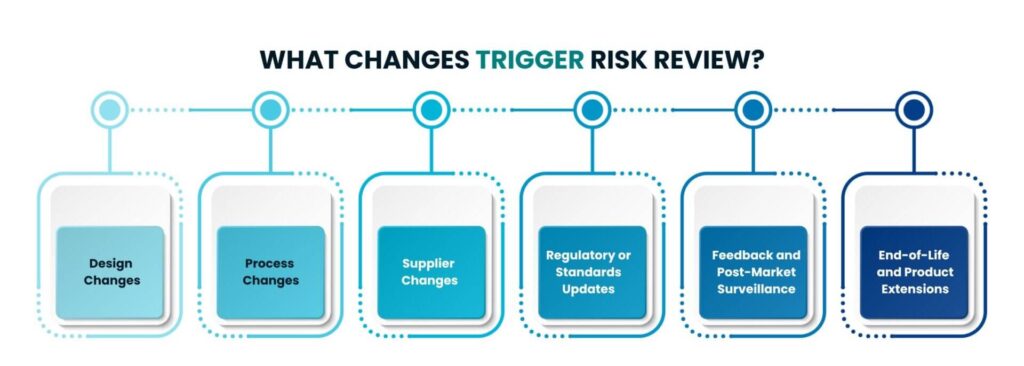
Following are the key scenarios that might prompt a reevaluation or update to a product’s risk management strategy, and calls for a risk review and update, as deemed necessary.
1. Design Changes:
Some common design changes that require a risk review via the FMEA tool are as follows:
⦿ Launching new features or functionalities.
⦿ Modifying existing designs, such as altering materials and components or updating software.
⦿ Integrating new technology or significantly redesigning the product.
Example of design change:
A medical device company manufactures an insulin pump and redesigns its casing to make it more durable and resistant to impact. The original casing was made of plastic, but the new design uses a metal alloy to enhance strength and durability.
Risk Review: Since this involves major design changes that can cause issues like overheating, increased device weight, and others, a risk review is necessary. The risk review might highlight critical areas that need attention due to the design change.
The company then should implement design adjustments, testing protocols, and quality checks to mitigate the identified risks. This ensures that the new, more durable insulin pump casing enhances the device’s reliability without compromising patient safety or device performance.
2. Process Changes:
The following process changes might trigger a risk review.
⦿ Introducing new manufacturing processes or automation.
⦿ Altering assembly lines, inspection procedures, or testing protocols.
⦿ Adjusting process parameters that could impact product quality.
Example:
A medical device company producing cardiac pacemakers decides to automate part of the assembly process to increase efficiency and consistency. The new process involves using robotic arms for the placement of sensitive electronic components, which was previously done manually by skilled technicians.
Risk Review: Since a significant process change happens here, a risk review is necessary to identify any potential risks associated with the new automated assembly procedure. Through the risk review, the company can detect issues like misalignment of electronic components, increased risk of component damage, etc.
If detected, by addressing these risks with targeted controls and monitoring systems, the company can benefit from increased production efficiency without compromising the safety or quality of the cardiac pacemakers. This proactive approach helps maintain product reliability and regulatory compliance as the new process is implemented.
3. Supplier Changes:
Mentioned below are some supplier changes that trigger risk review. Supplier changes may occur for addressing issues with supplier performance, like consistent delivery of nonconforming parts. Supplier changes include but are not limited to:
⦿ Switching to a new supplier for critical components or materials.
⦿ Changes in supplier processes that could affect the quality or reliability of parts.
Example:
Let’s consider a situation in which a medical device company manufactures an insulin pump and decides to switch to a new supplier for its main component, the pump’s microcontroller. The new microcontroller seems to have a different performance characteristic than the last one.
Risk Review: The change in characteristics of the new microcontroller calls for the risk review since there might be a possibility of new failure modes, like issues with signal processing, timing, and power consumption. Through the risk review, the team can detect issues like “delayed insulin delivery” or overheating. Following this analysis, they can then implement additional controls or testing procedures to mitigate the risk.
4. Regulatory or Standards Updates:
Below, are a few regulatory or standards updates that call for a risk review.
⦿ Updates to regulatory requirements or industry standards that impact product compliance or risk management.
⦿ New regulations requiring additional controls or risk assessments.
Example:
A medical device company that manufactures surgical instruments is informed of an update in sterilization standards by a major regulatory body. The new standards require a more stringent sterilization process to ensure that all instruments meet higher sterility assurance levels. The company must update its existing sterilization procedures to comply with these new regulatory requirements.
Risk Review: This regulatory requirement calls for risk review. By conducting a risk review, companies can detect potential risks associated with the sterilization process, such as inadequate sterilization process, increased wear on instruments, production delays, etc. The FMEA review ensures that the company addresses the risks associated with updating its sterilization process to meet the new regulatory standards. This careful approach helps avoid regulatory penalties and ensures the safety of end-users.
5. Feedback and Post-Market Surveillance:
Some feedback and post-market surveillance data also call for FMEA review. Some of these are as follows:
⦿ Significant customer complaints or adverse event reports indicating potential new risks.
⦿ Trends in postmarket surveillance data suggesting emerging issues.
⦿ Findings from internal audits, CAPA activities, or regulatory inspections.
Example:
Suppose a new surgical laser system is launched and the company has received multiple complaints about the device’s intermittently shutting down during procedure. After conducting internal audits and post-market surveillance, the company also detected the same issue.
Risk Review: Multiple complaints about the device calls for a risk review. It is important that the company reviews the FMEA and integrates this new feedback to identify the failure mode related to “unanticipated system shutdowns”. The company then needs to analyse the root cause and mitigate the issue.
6. End of Life and Product Extensions:
The following changes can trigger risk review.
⦿ Extending a product’s life beyond its originally planned duration.
⦿ Introducing new product variants or line extensions with the same design or manufacturing processes.
Example:
A medical device company has a popular vital signs monitor that is reaching the end of its product lifecycle. The company plans to phase out the current model and introduce an extended version with enhanced features, including improved data connectivity and a larger display. The transition requires careful management to ensure continued support for the existing product while launching the new version.
Risk Review: As the company plans the end-of-life (EOL) for the current model and the introduction of the extended version, an FMEA review is conducted to identify and mitigate potential risks during this transition. Some of these risks that can occur in this case are discontinuation of spare parts, compatibility issues, product launch delays, transition management challenges, etc. The FMEA review for the end-of-life of the current vital signs monitor and the introduction of the product extension helps the company proactively address potential risks.
What is FMEA?
The change management process is utilised in case of any major changes and most changes, as detailed above, do call for a period risk review.
The first step is to initiate a Change Request Form (CRF). This form should include a section that addresses whether the change impacts any existing failure modes, introduces any new ones, or requires changes to controls.
Followed by CRF is the step where impact analysis must be conducted. This includes a risk review to assess the changes in the risk profile of the overall product. There is a possibility that the risk profile gets impacted due to the changes introduced. Considering that additional measures must be implemented to mitigate any new or different risks that may be introduced.
Once the impact analysis is complete, the CRF and impact analysis are presented in a cross-functional meeting, where the change is discussed amongst the entire team, including representatives from engineering, manufacturing, regulatory, quality, and management.
Here, the presented changes can be approved or denied.
If the proposed changes are approved, they are implemented following the applicable procedures at the company and the updated product/process documentation is reviewed, approved, and signed off on.

What Happens when Risk is Impacted By The Introduced Change?
In case any change impacts the risk profile of a device, it is mandatory to consider a well-defined risk assessment criteria, defined below.

You must define clear criteria for assessing the level of risk that is associated with the change being introduced. In case the change meets or exceeds the set risk threshold, a detailed investigation must be triggered. You should make sure that no change is implemented until a detailed review has been completed. Doing this ensures safety and prevents any unintended consequences.
What happens if there are feedback based changes?

You must have a proper process in place to record and monitor customer feedback, complaints and postmarket surveillance data. If in any case the issue is reported that suggests a potential new failure mode or weakness in current controls, an FMEA review must be conducted.
NOTE: Internal Audits and Regulatory Inspections
You must regularly check the findings from internal audits and regulatory inspections. If you find any potential risk or noncompliance, an FMEA review must be triggered.
Documentation and Communication Changes

Document all Reviews
It is important that all the FMEA reviews and triggers that initiated the reviews are well documented. By doing so, a clear record of ISO 14971 risk management activities is present and compliance is ensured.
Communicate Changes
Having a proper chain of communication about FMEA updates is critical. You must communicate all these changes to the relevant stakeholders, including the production team, manufacturing team, regulatory team, or quality assurance team, to ensure that all changes are being properly implemented.
Conclusion
In this article, we have discussed everything from what risk and medical device risk analysis are to their importance. Not just that, we have also elaborated on the changes that trigger a periodic risk analysis and the procedures that need to be followed in such cases.
Finally, we would like to explain what can happen if the medical device risk review is not conducted periodically.
Not following proper risk review, when triggered, can lead to serious repercussions, including product failures, regulatory non-compliance, increased liability, safety hazards, and potential harm to users.
It can also damage a company’s reputation, result in costly recalls, and lead to legal action or financial losses.
In short, it is a critical process for any medical device company to emphasize on.
FAQs
What is required in ISO 14971 risk analysis?
The following things are required for risk analysis
⦿ Define the Intended Use and Reasonably Foreseeable Misuse
⦿ Identify Characteristics Related to Safety
⦿ Identify Known and Foreseeable Hazards
⦿ Estimate the Risk for Each Hazardous Situation
⦿ Document Assumptions and Data Sources
⦿ Maintain a Risk Analysis File
⦿ Use a Suitable Risk Analysis Method
What’s the difference between FMEA and FTA?
FMEA is a bottom-up analysis identifying failure modes and effects, while FTA is a top-down method that uses logic diagrams to trace system-level failures to root causes.
How is residual risk evaluated?
Residual risk is evaluated after risk control measures are applied. If the remaining risk is acceptable based on benefit-risk analysis, it can be justified with supporting data.
Do Class I devices require formal risk analysis?
Yes. While the depth of analysis may be lower, ISO 14971 applies to all device classes. Even Class I products need hazard identification and documented risk evaluations.
How often should risk files be updated?
Risk files should be updated when new information becomes available—such as during design changes, CAPA findings, complaint trends, or post-market data reviews.



















