
Introduction
The global medical device regulatory landscape has grown significantly more complex in 2026. Enforcement of the EU Medical Device Regulation (EU MDR) has tightened considerably since its 2021 application date, with staggered transition deadlines extending through 2027 and 2028 for certain device classes. Meanwhile, the FDA's transition to the Quality Management System Regulation (QMSR), effective February 2, 2026, has mandated alignment with ISO 13485:2016 standards. Add the expanding scope of the Medical Device Single Audit Program (MDSAP)—now spanning the US, Canada, Australia, Brazil, and Japan—and regulatory affairs has become one of the most resource-intensive functions for device manufacturers.
Most medical device companies—from early-stage startups to mid-sized manufacturers—now turn to outsourced regulatory affairs partners rather than building in-house teams. The global regulatory affairs outsourcing market reached $7.03 billion in 2024 and is forecast to hit $11.31 billion by 2030, growing at an 8.3% CAGR, with the medical device segment projected at 7.9% CAGR.
Outsourcing provides immediate access to regulatory expertise, reduces submission delays, and keeps companies audit-ready across multiple markets simultaneously, without the long-term cost of permanent headcount.
This article evaluates 10 leading outsourced regulatory affairs companies based on global reach, service depth, regulatory certifications, and demonstrated track record with medical device approvals in 2026 — so you can shortlist the right partner for your next submission or market expansion.
Key Takeaways
- Outsourcing regulatory affairs cuts time-to-market without building a permanent in-house team
- Top partners offer end-to-end support: 510(k)/PMA/EU MDR submissions, QMS, post-market surveillance
- Shortlist partners by regulatory body coverage, device specialization, and engagement flexibility
- Elexes, Emergo by UL, MCRA, Arazy Group, and ProPharma Group lead the 2026 rankings
- The right fit depends on device type, target markets, and development stage — not budget alone
Why Medical Device Companies Outsource Regulatory Affairs
Outsourced regulatory affairs means external firms take ownership of submissions, quality management system (QMS) compliance, audit preparation, and post-approval activities on behalf of the manufacturer. These partners function as an extension of the company's regulatory team, delivering expertise that would otherwise require years and considerable investment to build internally.
The 2024 RAPS Global Compensation and Scope of Practice Report puts the cost of in-house talent in sharp relief:
| Role | US Average Base Salary | EU Average |
|---|---|---|
| VP / Director | $218,941 | €189,714 |
| Consultant | $180,112 | — |
| Manager | $145,766 | €95,427 |

Building an in-house team means committing to these figures plus benefits, training, and turnover costs. For startups and mid-sized firms, that overhead is often prohibitive.
Three primary drivers push companies toward outsourcing in 2026:
Multi-Market Submission Complexity
Pursuing FDA clearance, EU MDR certification, and MDSAP compliance simultaneously requires expertise across divergent regulatory frameworks. The FDA's QMSR final rule now incorporates ISO 13485:2016 by reference, while the EU MDR demands technical documentation under Annex II, and MDSAP harmonizes audits for five participating countries. Managing these concurrently without specialized expertise creates submission delays and rejection risk.
Shortage of In-House Regulatory Talent
The RAPS report found that 76% of professionals entered the field specifically because of high demand — a clear sign of a tight hiring market. The typical regulatory professional carries 17 years of total experience and 9 years in regulatory affairs specifically. For companies without established regulatory departments, recruiting and retaining professionals at that level is both slow and expensive.
Cost of a Full Internal Team
Maintaining a full-time regulatory affairs director, manager, and specialist can exceed $500,000 annually in the US alone. Outsourcing converts this fixed cost into a variable expense, letting companies scale support on a project or retainer basis without long-term headcount commitments.
Top 10 Outsourced Regulatory Affairs Companies for Medical Devices 2026
Companies were selected based on regulatory body coverage, device-specific expertise, certifications, engagement flexibility, and client track record.
Elexes
Elexes is a specialized medical device regulatory and quality compliance consulting firm with 50+ years of collective team experience and a 90% audit clearance rate. The firm has completed 250+ successful projects for 100+ global clients across SaMD, IVDs, implants, wearables, combination devices, and companion diagnostics.
End-to-end project ownership is what separates Elexes from generalist consultancies. Coverage spans FDA (510(k), PMA, De Novo), EU MDR, EU IVDR, Health Canada, TGA, and MHRA — from product development through post-market surveillance. Engagement models run full-time, part-time, or project-based with no long-term commitment required. A dedicated regulatory due diligence function identifies compliance risks before they trigger rejections or delays.

| Key Services | Regulatory submissions (FDA, EU MDR, MDSAP), QMS support (ISO 13485, ISO 9001), clinical evaluation reports, regulatory due diligence, compliance audits, post-approval surveillance | | Markets Served | USA, Europe, UK, Canada, Australia, Saudi Arabia, Asia, Hong Kong | | Best For | Startups to mid-sized device manufacturers needing end-to-end regulatory support across multiple global markets with flexible engagement terms |
Emergo by UL
Emergo by UL is a globally recognized medical device and IVD regulatory consulting firm operating in 25+ countries, known for deep specialization in device registration, QMS compliance (ISO 13485), and regulatory strategy across FDA, EU MDR, and Asia-Pacific markets.
Proprietary digital tools set Emergo by UL apart: RAMS® handles regulatory intelligence tracking, while OPUS™ supports human factors engineering workflows. EU MDR technical file preparation is a core strength, backed by notified body relationships that can compress CE marking timelines. With offices in 25 countries and device registration support across 20+ markets, the firm suits companies managing multi-country submissions simultaneously.
| Key Services | Device registration, EU MDR/IVDR technical files, QMS implementation, regulatory strategy, CE marking | | Markets Served | USA, EU, Canada, Asia-Pacific, Latin America (25+ countries) | | Best For | Companies pursuing CE marking or multi-market registrations who value digital regulatory tracking tools |
MCRA
MCRA is a US-headquartered regulatory consulting firm that integrates regulatory strategy, clinical, and reimbursement services specifically for medical devices, offering one of the most comprehensive pre-market and post-market service models in the industry. MCRA became an IQVIA business in 2024, expanding its global advisory capabilities.
Few firms combine regulatory, clinical, and reimbursement strategy under one roof — MCRA does. That integration lets device companies address all market access barriers without juggling multiple vendors. Deep FDA CDRH expertise drives high submission success rates, and the 2022 acquisition of Vorpal Technologies K.K. added meaningful Japan coverage alongside its US and EU presence.
| Key Services | 510(k)/PMA/De Novo submissions, IDE applications, clinical strategy, reimbursement consulting, post-market surveillance | | Markets Served | USA, EU, Japan | | Best For | Device companies seeking a single partner for regulatory, clinical, and reimbursement strategy in the US market |
Arazy Group
Arazy Group is an international medical device and IVD regulatory consulting firm operating in 140+ countries, distinguished by its proprietary regulatory technology platforms LICENSALE® and REGISLATE® that automate and manage multi-country registration workflows.
With 140+ countries covered, Arazy Group has the widest geographic reach on this list. The LICENSALE® and REGISLATE® platforms automate multi-country registration workflows, cutting manual tracking errors and accelerating timelines. Authorized representative services (EU AR, UK REP, CH REP) are included, making Arazy a single point of contact for companies managing global market entry without building internal infrastructure.
| Key Services | Global device registration, regulatory lifecycle management, authorized representative services, regulatory intelligence | | Markets Served | 140+ countries globally | | Best For | Companies pursuing simultaneous multi-country registrations at scale who need digital regulatory management tools |
ProPharma Group
ProPharma Group is a large life sciences consulting firm with a dedicated medical device practice offering full lifecycle regulatory affairs, pharmacovigilance, and compliance consulting across the USA, Europe, and Asia-Pacific.
ProPharma Group's depth comes from scale — a large global bench of regulatory specialists means consistent bandwidth even on complex, multi-market projects. The team has proven capability responding to FDA information requests and managing post-approval compliance. For companies expanding internationally, regulatory documentation and local representation are available across FDA and global submission pathways.
| Key Services | Regulatory affairs, compliance consulting, pharmacovigilance, post-market surveillance, international market expansion support | | Markets Served | USA, EU, Asia-Pacific | | Best For | Mid-to-large device companies needing scalable regulatory resources across multiple markets and product lines |
Freyr
Freyr is a regulatory affairs services company covering 120+ countries, offering end-to-end regulatory support for medical devices and IVDs with a strong emphasis on regulatory technology and digital submission management.
Cost-effectiveness relative to global reach is Freyr's clearest differentiator. Regulatory content management tools keep multi-country dossiers organized, and the firm has deep experience with APAC and emerging market registrations that many US-focused consultancies underserve. Coverage extends to regulatory strategy, QMS development (ISO 13485, 21 CFR Part 820, MDSAP), post-market surveillance, and EU Authorized Representative services.
| Key Services | Device registration, regulatory dossier management, labeling compliance, regulatory intelligence, APAC market access | | Markets Served | 120+ countries including APAC, EU, US, Middle East | | Best For | Companies targeting cost-effective regulatory support across high-volume emerging markets and APAC registrations |
PharmaLex
PharmaLex is a specialized life sciences regulatory and quality consulting firm serving pharmaceutical, biotech, and medical device companies, known for flexible and tailored regulatory affairs outsourcing engagements.
PharmaLex punches above its weight on EU MDR and IVDR depth. Gap analysis, technical documentation, clinical and performance evaluations, and EUDAMED support are core offerings — not add-ons. Despite operating from 60+ offices across 32 countries, the firm maintains a personalized service model that feels more like a dedicated consultant than a large agency. QMS consulting covers ISO 13485, MDSAP, and 21 CFR 820, with inspection readiness built into engagements.
| Key Services | EU MDR/IVDR compliance, QMS consulting, regulatory strategy, product lifecycle management, clinical evaluation | | Markets Served | EU, USA, Canada, Asia | | Best For | Companies needing specialized EU MDR compliance support with personalized consultant engagement |
MWA Consulting
MWA Consulting is a GxP compliance and regulatory consulting firm with 300+ associates across the USA, Canada, Europe, Asia, and South America, offering regulatory support across medical device, pharma, and biotech sectors.
Cross-functional GxP coverage is MWA's core value — GMP, GCP, and GLP capability across multiple industries, paired with structured training programs that consulting firms often skip. MWA Consulting has a well-established track record in clinical quality assurance and FDA inspection readiness, making it a practical fit for device companies that need compliance support reaching beyond regulatory affairs alone.
| Key Services | GxP compliance, QMS building, 510(k) submissions, internal audits, clinical quality assurance, FDA inspection readiness | | Markets Served | USA, Canada, Europe, Asia, South America | | Best For | Device companies that also need cross-functional GxP compliance support or integrated clinical quality assurance programs |
Regulatory Compliance Associates (RCA)
Regulatory Compliance Associates (RCA) is a long-standing regulatory and quality consulting firm serving the medical device, pharma, and biotech industries with a strong track record in FDA compliance, quality system remediation, and global market access. RCA is part of Sotera Health and serves 50+ countries.
RCA's remediation track record is concrete: a Fortune 500 device company achieved zero FDA 483 observations on follow-up audit after RCA-led remediation. That outcome reflects the firm's focused capability in quality system gap assessments and corrective action planning. Former FDA officials on staff give RCA direct insight into how inspectors evaluate submissions and quality systems — a practical advantage during high-stakes compliance situations.
| Key Services | FDA compliance consulting, QMS remediation, regulatory submissions, audit support, gap assessments | | Markets Served | USA, EU, global markets | | Best For | Device companies dealing with FDA warning letters, 483 observations, or quality system remediation needs |
MDI Consultants
MDI Consultants is a global quality management and regulatory consulting firm with experience in medical devices, pharma, food, and biotech, offering FDA and ISO compliance consulting across US, Canada, and European markets.
MDI Consultants combines regulatory consulting with clinical trial management — a pairing that most firms on this list don't offer. Experience in CE marking technical file preparation and 510(k) preparation covers the standard pre-market needs, while crisis intervention capability for FDA 483 and audit responses makes MDI a useful resource when compliance issues escalate quickly.
| Key Services | FDA/ISO compliance, CE marking, 510(k) preparation, clinical trial management, IDE submissions, 483 crisis response | | Markets Served | USA, Canada, EU | | Best For | Device companies needing combined regulatory + clinical trial management support, or those in an active FDA compliance crisis |
How We Chose the Best Outsourced Regulatory Affairs Partners
Companies were assessed against criteria directly tied to the core challenges medical device manufacturers face when outsourcing—not just firm size or brand name. Choosing a partner on price alone — without verifying device-specific experience or regulatory body coverage — is the fastest path to a failed submission.
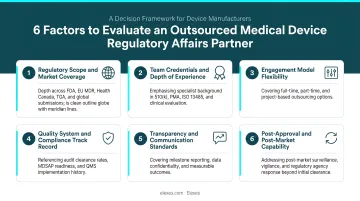
Key evaluation factors:
1. Regulatory Body Coverage Does the firm demonstrate proven expertise with FDA (510(k), PMA, De Novo), EU MDR/IVDR, Health Canada, TGA, and MDSAP? According to the FDA MDSAP program, participating regulatory authorities include the US (FDA), Canada (Health Canada), Australia (TGA), Brazil (ANVISA), and Japan (MHLW/PMDA). Firms should show active engagement with all relevant authorities for your target markets.
2. Device-Type Specialization Does the firm have documented experience with your specific device category—SaMD, IVDs, implants, wearables, combination devices, or companion diagnostics? Regulatory requirements vary significantly across device types. Broad consulting credentials don't substitute for category-specific submission history.
3. Engagement Model Flexibility Does the firm offer project-based, part-time, or ongoing support models that match your company's stage and resource constraints? Startups typically need flexible, lower-commitment arrangements; established manufacturers may prefer retainer-based support.
4. Certifications and Quality System Compliance Is the firm itself compliant with ISO 13485 or other quality standards? A consulting firm that maintains rigorous internal quality practices is better positioned to guide your compliance efforts.
5. Proven Track Record Can the firm demonstrate audit clearance rates, submission success rates, and documented client outcomes? Ask for named device categories and jurisdictions — not just client counts.
6. Post-Approval and Post-Market Surveillance Capability Does the firm support vigilance reporting, periodic safety update reports (PSURs), post-market clinical follow-up (PMCF), and complaint handling? Approval is a milestone, not a finish line — confirm the firm can support what comes after.
A startup preparing its first FDA 510(k) needs a fundamentally different partner than a multinational pursuing simultaneous EU MDR, MDSAP, and TGA submissions. Use these criteria to match on fit — not firm reputation.
Conclusion
The right outsourced regulatory affairs partner fits your specific regulatory pathway, device type, and development stage — not simply the firm with the largest global footprint. Evaluate ongoing performance indicators such as audit clearance rate, submission turnaround, and post-market surveillance capability before signing an agreement. These partnerships extend well beyond initial approval — choose a firm capable of supporting your device through its entire lifecycle.
Elexes brings 50+ years of collective expertise, a 90% audit clearance rate, and coverage across FDA, EU MDR, Health Canada, and more — from early development through post-market surveillance. Contact Elexes for a free initial consultation at +1 408-475-8091 or visit www.elexes.com/contact.
Frequently Asked Questions
What does an outsourced regulatory affairs company do for medical devices?
Outsourced regulatory affairs firms manage submissions (510(k), PMA, EU MDR, etc.), QMS compliance, audit preparation, and post-market surveillance on behalf of the manufacturer. They act as an extension of your internal team without requiring permanent headcount.
How much does it cost to outsource regulatory affairs for a medical device?
Costs depend on scope, target markets, and engagement model. Hourly rates typically run $150–$500, while project-based fees for deliverables like FDA 510(k) submissions or EU MDR technical files range from $15,000 to $150,000 depending on device complexity and clinical evidence requirements. See industry benchmarks for further detail.
What is the difference between a regulatory affairs consultant and an outsourced regulatory affairs company?
A consultant is an individual expert hired for specific tasks. An outsourced regulatory affairs company provides a full team covering submissions, QMS, audits, and clinical evaluation under a formal project management structure — suited for ongoing or multi-market needs.
How do I choose the right outsourced regulatory affairs partner for my medical device?
The most important factors are regulatory body coverage matching your target markets, proven experience with your specific device type, flexible engagement terms, and a verifiable track record including audit success rates and submission outcomes.
Can outsourced regulatory affairs companies handle both FDA 510(k) and EU MDR submissions?
Many firms on this list handle both, but quality of execution varies. Companies should specifically verify the firm's EU MDR technical documentation experience and notified body relationships, not just their stated geographic coverage.
Is outsourcing regulatory affairs a good option for medical device startups?
For most startups, outsourcing is the fastest path to regulatory expertise. You get immediate access to experienced professionals without building an in-house team, and many firms offer project-based or part-time models that match early-stage budgets and timelines.


