
The U.S. medical device manufacturers market was valued at $256.2 billion in 2024, growing at 5.9% annually toward a projected $360.1 billion by 2030. This scale makes it one of the most heavily regulated medical device markets globally. Regulatory consulting firms help companies navigate FDA submissions, build compliant Quality Management Systems (QMS), and secure global market access while avoiding the costly pitfalls of non-compliance.
This list profiles the top 10 medical device regulatory consulting firms in the USA for 2026, what makes each stand out, and how to choose the right partner for your needs.
Key Takeaways
- The right regulatory consulting firm can cut months off your FDA clearance timeline and prevent the submission errors that derail device launches
- Top firms are evaluated on regulatory expertise, FDA track record, service breadth, client size fit, and global reach
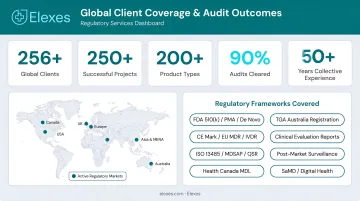
- Elexes leads for end-to-end regulatory support, 50+ years of collective experience, and 90% audit clearance rate across 250+ projects
- Choose based on your device class, target markets, and whether you need project-based help or ongoing outsourced support
- Specialists in your device type and regulatory pathway (510(k), PMA, De Novo, EU MDR) outperform generalists
What Is Medical Device Regulatory Consulting — and Why Does It Matter in 2026?
Medical device regulatory consulting provides specialized advisory services that help manufacturers navigate regulatory pathways (FDA, ISO 13485, EU MDR, Health Canada), build quality management systems, prepare submissions, and maintain post-market compliance.
2026 brings a wave of overlapping regulatory deadlines that companies can't afford to miss. Key changes include:
- FDA finalized PCCP guidance for AI-enabled devices in December 2024, followed by draft AI lifecycle management guidance in January 2025
- The Quality Management System Regulation (QMSR) takes effect in February 2026, replacing the legacy QSR
- EU MDR transition deadlines require Class III and most Class IIb implantable devices to achieve full compliance by December 31, 2027
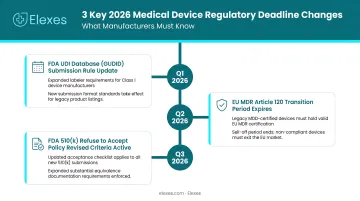
Each of these shifts requires a different compliance strategy. The firms below specialize in exactly that — helping companies map the right path, build the documentation, and cross the finish line without costly delays.
Top 10 Medical Device Regulatory Consulting Firms in the USA 2026
Each firm below was evaluated on regulatory expertise, FDA submission track record, service range, client outcomes, and the ability to serve companies from early-stage startups through enterprise scale. Use the breakdowns to match your device type, risk class, and target markets to the right partner.
Elexes Medical Consulting
Elexes is a globally recognized medical device regulatory consulting firm with 50+ years of collective team experience, 250+ completed projects across 200+ product types, and a 90% audit clearance rate. The firm serves 100+ global clients including AliveCor, DJO Global, Outset Medical, and Novasignal, offering end-to-end regulatory support from product development through post-market surveillance.
Key differentiators include:
- Cross-functional outsourcing that combines regulatory, quality, and clinical tasks under one roof
- Flexible engagement options: full-time, part-time, or project-based
- Coverage across FDA, ISO 13485, EU MDR, Health Canada, EMA, MHRA, and MDSAP
- Specialized due diligence team that identifies red flags before they delay approvals
| Category | Details |
|---|---|
| Core Services | Regulatory submissions (510(k), PMA, De Novo), QMS setup and support, clinical trial documentation, regulatory due diligence, post-market surveillance, MDSAP, ISO 13485 compliance |
| Key Regulatory Bodies | FDA, ISO 13485, ISO 14971, IEC 62304, EU MDR, Health Canada, EMA, MHRA, MDSAP |
| Best Suited For | Startups to enterprise medical device companies, IVD manufacturers, SaMD developers, wearables, implant manufacturers, digital health companies |
Emergo by UL
Headquartered in Austin, TX, Emergo by UL operates in 30+ countries and is known for its depth in FDA regulatory strategy, QMS compliance, and CE marking support. The firm differentiates through its integration with UL Labs, giving clients a unique edge in cybersecurity compliance, SaMD, and AI device regulatory strategy—areas of growing FDA scrutiny in 2026.
| Category | Details |
|---|---|
| Core Services | Regulatory strategy, device registration, ISO 13485 QMS, UDI implementation, human factors engineering, MDSAP, CE marking |
| Key Regulatory Bodies | FDA, EU MDR, Health Canada, ISO 13485, TGA, MDSAP |
| Best Suited For | Companies seeking global market entry, SaMD developers, Class II and III device manufacturers |
NAMSA
Based in Northwood, OH, NAMSA (North American Science Associates) is a leading Medical Research Organization (MRO) that uniquely combines regulatory consulting with in-house clinical research and biocompatibility testing labs (ISO 10993). Their integrated model—covering preclinical, clinical, and post-market phases under one provider—makes them especially strong for Class II and Class III device manufacturers that need both regulatory strategy and clinical trial management.
| Category | Details |
|---|---|
| Core Services | Biocompatibility testing, clinical trial management, regulatory submissions, FDA and CE compliance, post-market studies |
| Key Regulatory Bodies | FDA, EU MDR, ISO 10993, ISO 13485 |
| Best Suited For | Class II/III device manufacturers, combination product companies, companies needing clinical + regulatory under one roof |
MCRA (Medical Device Regulatory Advisors)
Based in Washington, DC, MCRA is built around a deep bench of former FDA reviewers and CMS advisors, giving clients direct insight into how regulators evaluate complex and high-risk device submissions. MCRA's ability to align regulatory, clinical, and reimbursement strategy simultaneously makes them a top choice for PMA submissions, breakthrough designation applications, and IDE studies—particularly in cardiovascular, neuromodulation, and orthopedic device categories.
| Category | Details |
|---|---|
| Core Services | PMA, IDE, De Novo, 510(k) submissions, reimbursement strategy, clinical trial design, breakthrough device designation |
| Key Regulatory Bodies | FDA (CDRH), CMS, EU MDR |
| Best Suited For | High-risk Class III device companies, established medtech firms, companies pursuing breakthrough device designation |
Regulatory Compliance Associates (RCA)
RCA is a US-based consultancy staffed heavily with former FDA officials and industry veterans, providing clients with deep insider knowledge of what regulators look for during submissions and inspections. RCA excels in compliance remediation—helping companies respond to FDA warning letters, restructure QMS systems, and conduct mock audits—making them a go-to when compliance issues have already surfaced and need expert resolution.
| Category | Details |
|---|---|
| Core Services | QMS development, mock audits, FDA warning letter response, CAPA management, regulatory submissions, risk management |
| Key Regulatory Bodies | FDA (21 CFR Part 820), ISO 13485, EU MDR |
| Best Suited For | Companies under FDA scrutiny, manufacturers needing remediation, firms preparing for FDA inspections |
MDI Consultants
MDI Consultants is a US-based firm with a strong track record in handling FDA 510(k), PMA submissions, and QSR compliance—particularly valued for their experience navigating FDA warning letters and enforcement actions. Their focused expertise in FDA compliance remediation and QMS restructuring makes them a strong choice for companies that have encountered regulatory challenges and need fast, experienced recovery support.
| Category | Details |
|---|---|
| Core Services | 510(k), PMA submissions, QSR compliance, FDA audit support, warning letter response, design controls |
| Key Regulatory Bodies | FDA (21 CFR Part 820), ISO 13485 |
| Best Suited For | Companies facing FDA enforcement actions, Class I–III manufacturers needing submission support |
RQM+
Headquartered in Monroeville, PA, RQM+ is a regulatory and quality consultancy known for EU MDR depth recognized for its EU MDR/IVDR gap assessments, clinical evaluation report writing, and expertise in biologics, combination products, and companion diagnostics. A deep bench of former notified body reviewers gives RQM+ a distinct advantage for companies preparing EU MDR technical files and post-market clinical follow-up documentation.
| Category | Details |
|---|---|
| Core Services | EU MDR/IVDR gap assessments, clinical evaluation reports, quality assurance, lab testing, post-market surveillance |
| Key Regulatory Bodies | EU MDR, EU IVDR, FDA, ISO 13485 |
| Best Suited For | Companies entering EU markets, combination product manufacturers, companion diagnostics developers |
Greenlight Guru
Based in Indianapolis, IN, Greenlight Guru occupies a unique niche as both a cloud-based QMS software provider and an integrated compliance consulting service—purpose-built for medical device companies navigating ISO 13485 and 21 CFR Part 820. Their software-plus-consulting hybrid model is particularly powerful for digital health and SaMD companies that need streamlined documentation, design controls, and audit readiness in a single platform.
| Category | Details |
|---|---|
| Core Services | QMS software implementation, design control documentation, ISO 13485 compliance, audit preparation, CAPA management |
| Key Regulatory Bodies | FDA (21 CFR Part 820), ISO 13485 |
| Best Suited For | Startups and digital health companies seeking tech-enabled QMS solutions, SaMD developers |
Medpoint
Medpoint is a US-based consultancy with a team of 350+ global consultants, offering one of the broadest service ranges in the industry—from QA support and FDA 21 CFR 820 compliance to clinical support, technical consulting, and global supplier auditing. Their scale and breadth of service coverage make them a strong choice for mid-to-large medical device companies that need a single consultancy partner to manage multiple regulatory workstreams simultaneously.
| Category | Details |
|---|---|
| Core Services | QA support, FDA and MDSAP compliance, clinical consulting, global supplier auditing, technical documentation |
| Key Regulatory Bodies | FDA (21 CFR Part 820), MDSAP, ISO 13485 |
| Best Suited For | Mid-to-large device manufacturers, companies with multi-market regulatory needs, firms requiring supplier audit support |
Freyr Solutions
Freyr Solutions operates across India and the USA, offering global regulatory submission capabilities powered by automation tools—supporting FDA, EU MDR, TGA, CDSCO, and UDI compliance across 120+ countries. Their investment in regulatory technology and automation gives cost-conscious companies a faster, more scalable path to multi-market submissions, making them particularly valuable for manufacturers targeting simultaneous global launches.
| Category | Details |
|---|---|
| Core Services | Global regulatory submissions, UDI compliance, labeling, regulatory automation, EU MDR, FDA 510(k) and PMA support |
| Key Regulatory Bodies | FDA, EU MDR, TGA, Health Canada, CDSCO, MDSAP |
| Best Suited For | Companies targeting multi-market global launch, cost-conscious manufacturers, firms needing regulatory automation |
How We Chose the Best Medical Device Regulatory Consulting Firms
Each firm on this list was evaluated against a consistent set of criteria — the same factors that matter when your submission timeline, clearance rate, or audit outcome is on the line.
Selection criteria included:
- Regulatory expertise across FDA, ISO 13485, and international bodies
- Service breadth from pre-submission through post-market surveillance
- Verified client outcomes, including audit clearance rates
- Flexibility to support both startups and established manufacturers
- Transparency in process, pricing, and measurable deliverables
- Ability to support you from early-stage development through product launch and beyond

Common selection mistakes to avoid:
- Choosing based on name recognition rather than device-class specialization
- Overlooking whether the firm has active FDA submission experience (not just theoretical knowledge)
- Failing to confirm engagement model fit — project-based, embedded, or retainer
Conclusion
The firms on this list cover a wide range of specializations in medical device regulatory consulting—but the right partner depends on your device type, risk class, target markets, and stage of development. Size and reputation matter less than fit.
When evaluating potential partners, look for:
- Direct experience with your specific device class and regulatory pathway
- A strong audit clearance track record with verifiable outcomes
- Flexible engagement models that scale with your stage of development
Elexes Medical Consulting provides end-to-end regulatory support across FDA, ISO 13485, EU MDR, and global markets, with 50+ years of collective expertise and a 90% audit clearance rate. To discuss your regulatory needs, reach out at +1 408-475-8091.
Frequently Asked Questions
What does a medical device regulatory consulting firm do?
These firms help medical device manufacturers navigate FDA submissions, build QMS systems, achieve ISO certifications, prepare for audits, and maintain ongoing post-market compliance.
How do I choose the right medical device regulatory consulting firm for my needs?
Evaluate firms based on their experience with your specific device class, familiarity with the target regulatory bodies (FDA, EU MDR, Health Canada), engagement model flexibility, and verified track record of successful submissions and audit clearances.
What is the difference between FDA 510(k) clearance and PMA approval?
510(k) is a premarket notification pathway for Class II devices that demonstrate substantial equivalence to a predicate device, while PMA (Premarket Approval) is a more rigorous pathway for Class III high-risk devices requiring clinical evidence of safety and effectiveness.
How much does medical device regulatory consulting typically cost?
Costs vary based on project scope, device class, and engagement model — from a few thousand dollars for document review to six figures for full regulatory submissions. Part-time, project-based, and full-outsourcing options allow companies at different budget levels to access expert support.
Do I need separate consultants for FDA and EU MDR compliance?
Most established firms cover both FDA and EU MDR under a single engagement, keeping technical documentation consistent and saving time. Still, verify the firm's active hands-on experience with each regulatory body rather than assuming broad coverage.
When is the right time to hire a medical device regulatory consultant?
Ideally, consultants are engaged during early product development to shape regulatory strategy from the start. Companies also bring them in at specific milestones — preparing FDA submissions, responding to warning letters, preparing for MDSAP audits, or planning international market entry.


