
Introduction
SaMD developers in the US face a compliance landscape that generalist medical device consultants are often unprepared for. FDA submissions now require navigating the Digital Health Center of Excellence, IEC 62304 software lifecycle processes, ISO 14971 risk management, evolving cybersecurity guidance, and AI/ML-specific frameworks simultaneously.
That regulatory environment demands specialized support. Generalist consultants frequently lack the software-specific depth needed to avoid costly FDA rejections or unnecessary submission delays.
The right SaMD regulatory consultant compresses time-to-market and manages your product lifecycle from pre-submission strategy through post-market surveillance. According to Mordor Intelligence's 2026 report, the global SaMD market is projected to grow from $5.24 billion in 2026 to $25.87 billion by 2031 at a 37.62% CAGR, with North America leading adoption.
That scale of growth has prompted the FDA to increase scrutiny on digital health submissions — making the choice of regulatory partner more consequential than ever.
This list covers the top 10 firms leading the US SaMD regulatory consulting space in 2026—vetted for SaMD-specific expertise, FDA submission track records, and demonstrated client outcomes.
Key Takeaways
- SaMD consultants handle FDA 510(k)/De Novo/PMA submissions, IEC 62304 compliance, and IMDRF risk classification — not just general device work
- The US SaMD market is growing at 37.62% annually, fueled by FDA's increased scrutiny of AI/ML devices and digital health software
- Vet firms on IEC 62304/ISO 14971 expertise, AI/ML submission track record, audit clearance rates, and engagement flexibility
- All 10 firms below are selected for SaMD-specific depth, US regulatory experience, and documented client outcomes
Overview of SaMD Regulatory Consulting in the US
SaMD regulatory consulting helps software-as-a-medical-device developers classify their products using the IMDRF's four-category risk framework, select the correct FDA submission pathway (510(k), De Novo, PMA, or exemption), and build compliant Quality Management Systems aligned with 21 CFR Part 820 and IEC 62304. These consultants bridge software development practices and medical device regulatory requirements — a combination that generalist device consultants rarely bring to the table.
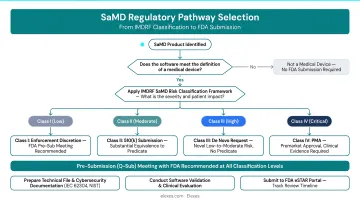
The 2026 US SaMD regulatory landscape is defined by several evolving frameworks:
- FDA's Digital Health Center of Excellence (DHCoE) coordinates digital health oversight across the agency and provides regulatory guidance, though it does not make final marketing authorization decisions
- AI/ML Action Plan implementation continues through new guidances, including the August 2025 final guidance on Predetermined Change Control Plans (PCCPs) for adaptive algorithms
- Updated Quality System Regulation (QMSR) took effect February 2, 2026, incorporating ISO 13485:2016 by reference and applying to all finished device manufacturers, including SaMD companies
This regulatory complexity explains why generalist consultants rarely have the depth SaMD companies need. The following 10 firms have been selected based on SaMD regulatory depth, US FDA experience, technical certifications, and client outcomes—giving you a shortlist of firms equipped to handle the specific demands of software-defined medical products in 2026.
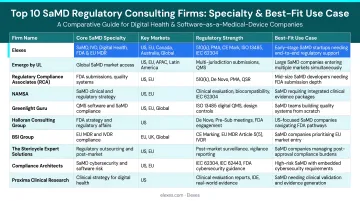
Top 10 SaMD Regulatory Consultants in the US 2026
Selection is based on demonstrated SaMD expertise, FDA submission track records, IEC 62304 and AI/ML capabilities, client base diversity, and engagement flexibility.
Elexes
Elexes is a US-based medical device regulatory consulting firm with 50+ years of collective experience, serving 100+ global clients across 200+ product types including SaMD, digital health, and AI-driven devices. Notable clients include AliveCor, Radformation, and Novasignal.
Elexes stands out for its end-to-end SaMD project management—from IEC 62304-compliant software development documentation through FDA submission, QMS implementation, and post-market surveillance. The firm reports a 90% audit clearance rate and offers flexible full-time, part-time, or project-based engagement models tailored to client stage and budget.
| Key SaMD Services | Relevant Certifications & Standards | Best Suited For |
|---|---|---|
| FDA 510(k)/De Novo support, IEC 62304 documentation, ISO 14971 risk management, QMS setup, MDSAP, clinical trial documentation, post-market surveillance | ISO 13485, IEC 62304, ISO 14971, MDSAP, HIPAA, GCP, GLP | SaMD startups to established digital health companies needing full-lifecycle regulatory support across FDA, EU MDR, Health Canada, and other global markets |
Emergo by UL
Emergo by UL is a global regulatory consulting firm with deep expertise in FDA, CE marking, Health Canada, and 30+ market entry pathways. The firm offers services tailored to SaMD, AI-driven devices, and cybersecurity compliance.
Emergo's integration with UL Labs gives clients access to combined regulatory strategy, cybersecurity assessments, human factors testing, and SaMD-specific UDI implementation through a single engagement. That consolidation is particularly useful for SaMD submissions where cybersecurity and human factors reviews run in parallel.
| Key SaMD Services | Relevant Certifications & Standards | Best Suited For |
|---|---|---|
| Regulatory pathway planning, SaMD classification, ISO 13485 QMS, cybersecurity consulting, human factors, UDI implementation | ISO 13485, MDSAP, FDA 21 CFR Part 820, IEC 62304 | Companies seeking multi-market SaMD entry with integrated cybersecurity and AI/ML regulatory support |
MCRA (Medical Device Regulatory Advisors)
MCRA is known for its bench of former FDA reviewers and CMS advisors, offering end-to-end regulatory strategy, reimbursement planning, and clinical trial management with growing specialization in digital health and SaMD pathways.
What sets MCRA apart is its ability to align FDA regulatory strategy with reimbursement planning within the same engagement. For SaMD companies building a commercial case alongside their submission dossier, that means payer coverage questions get addressed before—not after—clearance.
| Key SaMD Services | Relevant Certifications & Standards | Best Suited For |
|---|---|---|
| FDA 510(k)/De Novo/PMA submissions, IDE and Breakthrough Device applications, digital health regulatory strategy, reimbursement alignment | FDA 21 CFR Part 820, De Novo, PMA pathways | SaMD developers needing combined regulatory and payer strategy, especially for Class II/III software |
RQM+
RQM+ is a full-service regulatory and quality consulting firm with a deep bench of former notified body reviewers and FDA regulatory experts, specializing in SaMD, companion diagnostics, biologics, and combination products.
RQM+ is particularly strong on EU MDR/IVDR gap assessments run in parallel with US FDA work—a practical advantage for SaMD companies that can't afford sequential market entry timelines.
| Key SaMD Services | Relevant Certifications & Standards | Best Suited For |
|---|---|---|
| SaMD regulatory submissions, clinical evaluation reports, EU MDR/IVDR compliance, post-market surveillance, quality assurance | ISO 13485, IEC 62304, EU MDR, IVDR | SaMD companies targeting both US and EU markets, including companion diagnostics and combination products |
Regulatory Compliance Associates (RCA)
RCA (part of the Sotera Health group) provides regulatory, quality, and compliance support for SaMD, AI/ML devices, and cybersecurity, drawing on subject matter experts experienced in non-traditional submissions.
RCA is known for its multidisciplinary approach to SaMD/AI submissions, including cybersecurity documentation, pre-submission meeting strategy, and remediation services for companies that have received FDA Warning Letters.
| Key SaMD Services | Relevant Certifications & Standards | Best Suited For |
|---|---|---|
| SaMD FDA strategy, AI/ML submission support, cybersecurity compliance, QMS remediation, pre-submission meeting preparation | ISO 13485, ISO 9001, FDA 21 CFR Part 820, 21 CFR Part 11 | SaMD and AI/ML device companies needing FDA regulatory strategy with cybersecurity integration, including remediation scenarios |
NAMSA
NAMSA is a full-service Medical Research Organization (MRO) headquartered in Northwood, OH, offering integrated regulatory consulting, biocompatibility testing, and clinical trial management—increasingly active in SaMD and digital therapeutics regulatory support.
NAMSA stands out by combining consulting with in-house lab capabilities for ISO 10993 biocompatibility and analytical chemistry, making it particularly valuable for SaMD products integrated with hardware (combination products).
| Key SaMD Services | Relevant Certifications & Standards | Best Suited For |
|---|---|---|
| Regulatory affairs, QMS development, clinical trial management, preclinical/post-market studies, combination product support | ISO 13485, ISO 10993, FDA 21 CFR Part 820, EU MDR | SaMD companies with hardware integration or combination product components needing clinical and regulatory services under one roof |
Sunstone Pilot Group
Sunstone Pilot specializes in SaMD, digital therapeutics, and connected device consulting with deep expertise in design controls, human factors engineering, and real-world evidence integration specific to software-based devices.
Sunstone's work is scoped exclusively to software-first medical devices—hands-on support for IEC 62304 documentation, failure mode analysis (FMEA), usability validation, and FDA pre-submission preparation without the generalist overhead of larger firms.
| Key SaMD Services | Relevant Certifications & Standards | Best Suited For |
|---|---|---|
| SaMD design controls, IEC 62304 documentation, human factors/usability validation, FMEA, risk management, FDA pre-submission support | IEC 62304, ISO 14971, FDA 21 CFR Part 820 | SaMD and digital therapeutics startups requiring hands-on design control and usability validation support |
Greenlight Guru
Greenlight Guru offers a hybrid consulting and software model—combining a cloud-based, FDA-compliant QMS platform purpose-built for medical devices with live consulting services for QMS implementation and audit preparation, including SaMD-specific workflows.
The platform-plus-consulting model reduces documentation burden for SaMD teams by embedding ISO 13485 and 21 CFR Part 820 compliance directly into day-to-day workflows, improving traceability and audit readiness without manual overhead.
| Key SaMD Services | Relevant Certifications & Standards | Best Suited For |
|---|---|---|
| QMS implementation, ISO 13485 compliance, design control documentation, audit preparation, SaMD-specific workflow consulting | ISO 13485, FDA 21 CFR Part 820 | SaMD companies wanting to streamline documentation and QMS management through a software-integrated consulting approach |
QES Medical
QES Medical is a boutique consulting firm with 20+ years of leadership experience in medical device regulatory affairs, offering targeted SaMD, QMS, and FDA compliance services to both device and pharma companies.
QES differentiates through personalized, senior-led engagements covering SaMD regulatory submissions, risk management, FDA compliance remediation, and ISO 13485 QMS building—making it a strong fit for small-to-mid-size SaMD teams needing focused expert access.
| Key SaMD Services | Relevant Certifications & Standards | Best Suited For |
|---|---|---|
| SaMD regulatory submission support, QMS building, FDA compliance remediation, risk management, ISO 13485 audits | ISO 13485, FDA 21 CFR Part 820 | Small to mid-size SaMD companies needing personalized, senior-level regulatory guidance without large firm overhead |
Orthogonal
Orthogonal is a Chicago-based software development and consulting firm exclusively focused on SaMD, providing full-scope services from UX design, IEC 62304-compliant development, risk analysis, and verification and validation for Class I through Class III software-based devices.
Orthogonal differentiates as the only firm on this list that combines SaMD engineering services with regulatory compliance, making it uniquely suited for companies that need both a development partner and a regulatory consultant embedded in one team.
| Key SaMD Services | Relevant Certifications & Standards | Best Suited For |
|---|---|---|
| IEC 62304 development documentation, risk analysis (ISO 14971), V&V protocols, SaMD design for Class I–III, mobile/web/cloud SaMD builds | IEC 62304, ISO 14971, FDA 21 CFR Part 820 | SaMD companies needing both software development and regulatory compliance expertise in a single partner |
How We Chose the Best SaMD Regulatory Consultants
This list focuses specifically on SaMD regulatory expertise—not general medical device consulting. Firms were evaluated on:
- Demonstrated SaMD submission experience across FDA 510(k), De Novo, and PMA pathways
- Knowledge of IEC 62304 software lifecycle processes and IMDRF SaMD classification
- AI/ML regulatory experience, including familiarity with FDA's AI/ML Action Plan and PCCP guidance
- Ability to handle the full SaMD product lifecycle from pre-submission through post-market surveillance
Choosing a generalist consultant without SaMD-specific FDA experience is one of the fastest ways to trigger an FDA RTA (Refuse to Accept) or waste months on preventable re-work.
Beyond core SaMD competency, each firm was also assessed on operational depth:
- Certifications held: ISO 13485, IEC 62304, MDSAP recognition
- Client diversity across SaMD categories: AI/ML, digital therapeutics, wearables, IVD software
- Audit clearance rates and track record of first-pass FDA acceptance
- Flexibility of engagement: project-based, retainer, or outsourced regulatory department models
- Global regulatory reach for companies targeting both US and EU markets

Conclusion
A qualified SaMD regulatory consultant reduces regulatory risk, accelerates FDA clearance timelines, and ensures your software lifecycle documentation—per IEC 62304 and ISO 14971—holds up under scrutiny. With the FDA actively updating guidance on AI/ML devices, cybersecurity, and Predetermined Change Control Plans, that specialization directly affects submission outcomes.
Given that context, who you hire matters beyond general medical device credentials. When evaluating consultants, assess their specific SaMD experience and familiarity with the FDA's current digital health priorities—the AI/ML action plan, cybersecurity guidance, and the Digital Health Center of Excellence's evolving frameworks. Also confirm their engagement model fits your stage and budget, whether you're a pre-revenue startup or an established company expanding your portfolio.
For SaMD developers needing support from development documentation through FDA submission and post-market surveillance, Elexes brings 50+ years of collective expertise, ISO 13485 and IEC 62304-aligned processes, and a 90% audit clearance rate across 250+ completed projects. Reach their team at +1 408-475-8091 to discuss your regulatory needs.
Frequently Asked Questions
What is a SaMD regulatory consultant and what do they do?
SaMD regulatory consultants help software-as-a-medical-device developers navigate FDA classification, select the right submission pathway (510(k), De Novo, or PMA), build IEC 62304-compliant documentation, and manage QMS and post-market requirements specific to software-based devices.
What FDA regulations apply to Software as a Medical Device in the US?
The FDA's Digital Health Center of Excellence oversees SaMD regulation. Key applicable standards and frameworks include:
- IMDRF SaMD framework
- 21 CFR Part 820 (now QMSR, incorporating ISO 13485)
- IEC 62304 (software lifecycle)
- ISO 14971 (risk management)
- FDA's AI/ML-Based SaMD Action Plan for adaptive algorithms
How long does it typically take to get FDA clearance for a SaMD product?
Timelines vary by submission type. Under MDUFA V performance goals, FDA targets 90 FDA Days for 510(k) decisions and 150 FDA Days for De Novo requests — though total calendar time typically exceeds 128 days when Additional Information Requests are factored in. Early pre-submission meetings and complete documentation packages help shorten this timeline.
What certifications should a SaMD regulatory consultant have?
Key certifications include ISO 13485, IEC 62304, ISO 14971, and MDSAP, with demonstrated experience in FDA digital health submissions. Membership in RAPS (Regulatory Affairs Professionals Society) and RAC certification are strong indicators of expertise.
What is the difference between SaMD and SiMD?
SaMD (Software as a Medical Device) is software intended to perform a medical function independently, while SiMD (Software in a Medical Device) is embedded software that operates as part of a physical device. The regulatory pathway and documentation requirements differ between the two, with SaMD requiring standalone validation under IEC 62304.
Do SaMD startups need a regulatory consultant?
Not legally required, but most startups benefit significantly from one. A consultant helps define regulatory strategy early (classification, submission type, QMS setup), prevents costly rework, and ensures documentation meets FDA standards before submission — reducing the risk of rejection or delays.


