
Introduction
The U.S. medical device market operates under some of the world's most stringent regulatory oversight, with the FDA 510(k) clearance pathway serving as the entry point for the majority of Class II devices. For manufacturers, selecting the right consulting partner is often the difference between smooth market entry and costly delays or rejections.
FDA data shows the average 510(k) now requires 1.58 review cycles, and most submissions receive at least one Additional Information request. An experienced consultant reduces those cycles by catching submission errors early, accelerating clearance timelines, and keeping manufacturers out of regulatory holds.
This guide profiles the top 10 FDA 510(k) consultants in the United States for 2026, along with evaluation criteria to help you select the right partner for your device.
Key Takeaways
- The 510(k) process demands demonstrating substantial equivalence to a predicate device—a technically complex, document-heavy process
- Specialized consultants reduce submission errors, shorten review timelines, and improve clearance rates
- Top consultants are evaluated on 510(k) track record, device type expertise, FDA communication capabilities, and end-to-end service coverage
- Elexes brings 50+ years of collective experience, 250+ completed projects, and coverage across 200+ medical device product types
What Is the FDA 510(k) Process and Why Do Companies Hire Consultants?
A 510(k) is a premarket submission to the FDA demonstrating that a device is substantially equivalent to a legally marketed predicate device in both intended use and technological characteristics. According to FDA guidance, this pathway is required for most Class II and some Class I and III devices before commercial distribution.
The process is technically demanding. Manufacturers must:
The process is technically demanding. Before a submission reaches FDA reviewers, manufacturers must:
- Identify suitable predicate devices with matching intended use and technological characteristics
- Compile performance testing data aligned to applicable standards
- Prepare detailed technical documentation across multiple sections
- Ensure labeling compliance with current FDA requirements
- Navigate shifting guidance documents and anticipate reviewer expectations
The complexity shows in the numbers. FDA received 4,049 510(k) submissions in fiscal year 2025, a 12.8% increase from the prior year. While the FDA's review clock averages 71.98 days, total elapsed time—including industry response to Additional Information requests—averaged 117.27 days in FY 2025. With 1.58 average review cycles, most submissions require at least one round of back-and-forth with the FDA.
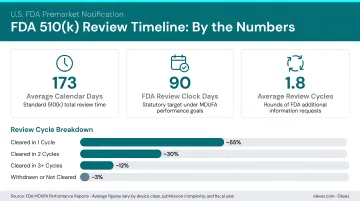
Consultants reduce that friction directly: they bring regulatory strategy expertise, cut down Additional Information (AI) requests through submission-ready documentation, and improve the odds of first-pass acceptance. The Refuse to Accept (RTA) rate dropped from 20.54% to 7.44% following the mandatory eSTAR electronic submission format—but even that means the FDA rejects roughly 1 in 14 submissions before review even begins.
The firms below have built track records helping medical device companies navigate exactly these challenges.
Top 10 FDA 510(k) Consultants in the United States (2026)
These firms were selected based on 510(k)-specific expertise, track record, device type coverage, client outcomes, and service comprehensiveness. Use the tables under each entry to quickly compare services, credentials, and fit for your device type.
Elexes
Elexes is a medical device regulatory consulting firm with 50+ years of collective experience, 250+ successful projects across 200+ product types, and a 98% first-pass acceptance rate for FDA 510(k) submissions.
The firm serves clients from startup medical device companies to large global manufacturers, covering FDA 510(k) submissions, QMS support, clinical trial documentation, regulatory due diligence, and post-market surveillance.
Elexes stands out for its end-to-end project ownership—from product development stage through 510(k) clearance to post-launch compliance—combined with flexible engagement models (full-time, part-time, or project-based) and complete data confidentiality. Notable clients include AliveCor, DJOGlobal, Outset Medical, and Radformation.
| Attribute | Details |
|---|---|
| Services Offered | 510(k) strategy & submission, predicate device identification, QMS (ISO 13485), clinical documentation, regulatory due diligence, post-market surveillance |
| Key Credentials | ISO 13485, ISO 14971, IEC 62304, MDSAP, GCP, GLP; expertise across FDA, EU MDR, Health Canada, MHRA, EMA |
| Best Suited For | Medical device companies, SaMD developers, IVD/LDT manufacturers, implant manufacturers, wearables, combination devices, and global manufacturers entering the U.S. market |
Emergo by UL
Emergo by UL is a specialist medical device and IVD regulatory consulting firm with a global office network spanning 20+ locations on six continents. Founded in 1997, the firm focuses exclusively on device regulatory affairs, quality assurance, and market access across the U.S., EU, Canada, and Asia-Pacific markets.
Emergo concentrates entirely on device classification, 510(k) premarket notification, and ISO 13485 QMS implementation. That narrow focus means consultants bring device-specific regulatory knowledge rather than generalist compliance support.
| Attribute | Details |
|---|---|
| Services Offered | 510(k) submissions, device classification, ISO 13485 QMS, IVD regulatory affairs, global market access |
| Key Credentials | ISO certified; part of UL Solutions with Notified Body and MDSAP services (kept separate to avoid conflicts) |
| Best Suited For | Mid-to-large medical device and IVD manufacturers seeking global market access |
FDA Group
Founded by former FDA professionals and based in Boston, FDA Group specializes in 510(k) submissions, qualification reviews, clinical operations support, and regulatory consulting with legal advisory capabilities. Their team's direct agency background provides practical insight into how reviewers evaluate submissions.
Their deep FDA connections directly benefit clients navigating pre-submission meetings, Additional Information requests, and interactive review phases. The firm reports a 97% client satisfaction rate and hundreds of 510(k) submissions completed.
| Attribute | Details |
|---|---|
| Services Offered | 510(k) submissions, regulatory strategy, clinical operations, FDA interaction management, consulting and legal advisory |
| Key Credentials | Team of former FDA officials and regulatory experts; strong FDA liaison experience |
| Best Suited For | Companies dealing with complex FDA interactions or requiring legal and regulatory advisory together |
EMMA International
EMMA International is a global consulting firm serving the medical device, pharmaceutical, and biotechnology sectors, with focused capability in quality regulatory services and timely regulatory document submission. Founded in 2012, the firm operates across eight industry sectors with headquarters in Birmingham, Michigan.
EMMA's strength lies in ensuring clients achieve market access without regulatory penalties or delays. Their team helps clients meet all applicable FDA requirements from device classification through post-clearance compliance.
| Attribute | Details |
|---|---|
| Services Offered | Regulatory submissions, quality management, FDA compliance, device classification, post-market support |
| Key Credentials | Multi-industry regulatory expertise; quality system implementation experience |
| Best Suited For | Medical device, pharma, and biotech companies needing integrated quality and regulatory support |
MDI Consultants
MDI Consultants has operated since 1978, when founder Alan Schwartz left the FDA to start the firm—giving it one of the longest track records in device and pharmaceutical regulatory consulting. The firm has served over 1,000 clients and reports completing over 2,000 combined 510(k)s, PMAs, and ANDAs.
Their team of former quality management leaders delivers practical, senior-level regulatory strategy backed by decades of cross-industry precedent. The firm maintains offices in the U.S., Europe, and Asia.
| Attribute | Details |
|---|---|
| Services Offered | 510(k) submissions, quality systems, regulatory strategy, compliance support for devices and pharma |
| Key Credentials | Operating since 1978; staff includes former FDA officials and ISO lead assessors |
| Best Suited For | Established device manufacturers looking for senior-level, experienced regulatory guidance |
Lachman Consultants
Lachman Consultants serves pharmaceutical, biotechnology, and medical device industries, offering compliance services including FDA inspection support, regulatory submissions, and technical training on regulatory processes. Founded in 1978, the firm is based in the New York area and serves global markets.
Lachman differentiates through its combination of regulatory submission support and practical staff training programs—helping client teams internalize FDA requirements, not just comply with them during engagement.
| Attribute | Details |
|---|---|
| Services Offered | 510(k) submissions, FDA inspection support, compliance consulting, regulatory training courses |
| Key Credentials | Multi-decade experience in pharma and device compliance; known for inspection readiness |
| Best Suited For | Companies needing both regulatory submission support and internal compliance capability building |
EAS Consulting Group
EAS Consulting Group is a regulatory firm with a network of over 200 independent advisors covering food, pharmaceutical, dietary supplement, and medical device sectors. This large advisory pool allows them to rapidly assign domain-specific experts to 510(k) projects.
Their broad consultant network is particularly valuable for novel device types or cross-category products that need multi-disciplinary regulatory input, ensuring the right expertise matches each engagement.
| Attribute | Details |
|---|---|
| Services Offered | Regulatory advisory, 510(k) support, auditing services, compliance solutions across FDA-regulated industries |
| Key Credentials | 200+ independent advisors; broad multi-sector regulatory coverage |
| Best Suited For | Companies with novel devices or cross-industry products needing specialized expert matching |
Ken Block Consulting
Founded in 2005 by Kenneth L. Block, Ken Block Consulting provides regulatory services exclusively to medical device companies, with offices in the U.S., EU, and Japan. The firm covers device classification, FDA submissions, product listings, and labeling compliance.
Their global footprint and focus on FDA agency interactions—including pre-submission meetings, marketing applications, and inspection support—make them a strong choice for manufacturers requiring coordinated U.S. and international regulatory strategy.
| Attribute | Details |
|---|---|
| Services Offered | Device classification, 510(k) submissions, product listings, FDA interaction management, labeling compliance |
| Key Credentials | Offices in U.S., EU, and Japan; RAC-certified founder with 35+ years experience |
| Best Suited For | Medical device companies pursuing parallel U.S. and international market entry |
The Weinberg Group (now ProPharma)
The Weinberg Group, now integrated into ProPharma, is a regulatory and compliance consultancy serving biotech and medical device companies, with capabilities in regulatory submissions including 510(k) premarket notifications and more complex PMA applications. The firm operates with global reach, including UK headquarters.
Their proximity to and established relationship with FDA's regulatory ecosystem, combined with scientific and legal advisory services, makes them a strong option for devices in complex or contested regulatory categories.
| Attribute | Details |
|---|---|
| Services Offered | 510(k) and PMA submissions, regulatory strategy, scientific advisory, compliance support |
| Key Credentials | Over 25 years in operation; combined scientific and regulatory expertise; strong FDA ecosystem connections |
| Best Suited For | Biotech and device companies with complex classification scenarios or high-risk product pathways |
Provision Consulting Group
Provision Consulting Group has served the medical device, food, and beverage industries with tailored compliance services including FDA registration, recall notifications, quality system training, and program audits. Based in Chino Hills, California, the firm reports over 2,200 global clients and 50,000+ projects completed.
Their emphasis on customized quality system programs ensures clients not only achieve 510(k) clearance but maintain post-market compliance standards that protect their market position. The firm is an Amazon SPN Partner for FDA Compliance.
| Attribute | Details |
|---|---|
| Services Offered | FDA registration, quality system training, program audits, recall notifications, compliance consulting |
| Key Credentials | Amazon SPN Partner; employs ex-FDA officials; multilingual staff |
| Best Suited For | Device manufacturers prioritizing post-clearance quality system strength and ongoing FDA compliance |
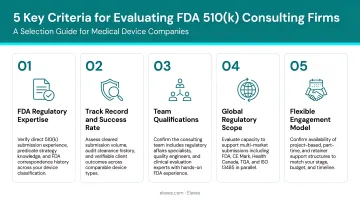
How We Chose the Best FDA 510(k) Consultants
Firms were assessed on specific 510(k) submission experience—not general regulatory consulting—along with device type coverage, evidence of successful clearances, FDA communication track record, and end-to-end service range.
A common mistake companies make is selecting general FDA consultants rather than firms with hands-on 510(k) and predicate strategy experience.
Key selection factors and their business outcomes:
- 510(k) track record: Submission quality and first-pass success rates matter. Firms with measurable outcomes—such as Elexes' 98% first-pass acceptance or FDA Group's hundreds of cleared submissions—demonstrate proven execution.
- Device type specialization: Your consultant should understand your device's specific risk class and testing requirements. Firms with exclusive medical device focus (like Ken Block Consulting) differ meaningfully from multi-sector firms like EAS or EMMA.
- FDA interaction capability: When additional information (AI) requests and review holds arise, turnaround speed depends on experience. Firms with former FDA reviewers (FDA Group, MDI Consultants) bring direct insight into reviewer expectations.
- Certifications and quality credentials: ISO 13485, ISO 14971, and RAC certification signal systematic, defensible regulatory practice aligned with QMS requirements.
- End-to-end support: Handoff risk between development, submission, and post-market stages is real. Elexes' project ownership from development through post-clearance demonstrates this approach.
Company size alone is not a reliable selection criterion. Boutique firms with specialized 510(k) focus often outperform larger generalist firms for device-specific submissions—demonstrated outcomes and submission track records matter more than brand recognition.
Conclusion
Choosing the right FDA 510(k) consultant shapes your regulatory pathway from predicate selection through clearance and beyond. A firm with proven experience in your specific device category can cut clearance timelines and reduce submission risk in ways a generalist simply cannot.
Before committing, evaluate consultants on more than hourly rates or firm size. The criteria that matter most:
- Demonstrated clearance history in your device category
- Transparent project timelines with defined milestones
- Scalability to support post-clearance compliance needs
- Strategic predicate selection experience — not just submission assembly
Submission quality and predicate strategy in the planning phase are what separate a 3-month clearance from a 12-month cycle of FDA deficiency responses.
For medical device companies that need a full-scope 510(k) partner, Elexes brings 50+ years of collective expertise, 250+ completed projects across 200+ device types, and a 90% audit clearance rate. Reach out to discuss your submission at +1 408-475-8091.
Frequently Asked Questions
Which consulting service is best for FDA medical device approval?
The best choice depends on device class, complexity, and submission type. Firms like Elexes that specialize in end-to-end 510(k) strategy, predicate identification, and FDA communication tend to deliver the strongest outcomes for Class II medical device clearances, particularly when they demonstrate measurable first-pass acceptance rates.
What does an FDA 510(k) consultant do?
A 510(k) consultant handles regulatory strategy, predicate selection, full submission preparation, and FDA communications including Additional Information requests. The best firms also support post-clearance compliance and reduce timelines through accurate, complete documentation.
How long does the FDA 510(k) clearance process take?
The FDA's standard review target is 90 days, and CDRH averaged 71.98 FDA days in FY 2025. However, total elapsed time including preparation and industry response to AI requests averaged 117.27 days in FY 2025. Full timelines from preparation through clearance typically range from 3-12+ months depending on device complexity and submission quality.
How much does it cost to hire an FDA 510(k) consultant?
Costs vary based on device complexity, submission type (Traditional, Special, or Abbreviated 510(k)), and scope of engagement. Full-service firms with former FDA reviewers or specialized device expertise typically command higher rates than limited-scope providers.
Do I need a 510(k) consultant if I already have an internal regulatory team?
Even experienced internal teams benefit from consultants on complex submissions or novel device categories. Consultants provide current insight on FDA reviewer preferences, up-to-date guidance knowledge, and surge capacity when responding to tight Additional Information request deadlines.
What is the difference between a 510(k) and a PMA submission?
A 510(k) is a premarket notification used for Class II devices demonstrating substantial equivalence to a predicate device. A PMA (Premarket Approval) is the most stringent FDA device marketing application, required for Class III high-risk devices, and demands clinical trial evidence demonstrating independent safety and effectiveness. The consulting requirements, timelines, and costs for a PMA are significantly higher than for 510(k) submissions.


