
Introduction
The European medical device market represents a substantial opportunity for US manufacturers, valued at approximately €160 billion in 2023 and accounting for 26.1% of the global market. The US is Europe's dominant trade partner, receiving 40.4% of EU medtech exports and supplying 45.4% of EU imports. However, this opportunity carries a significant regulatory price: EU MDR (Regulation (EU) 2017/745) replaced the Medical Device Directive (MDD) in 2021 with substantially stricter requirements that have fundamentally transformed European medical device compliance.
US companies entering the EU market face a distinct set of hurdles with no domestic equivalent:
- Appointing an EU Authorized Representative
- Preparing rigorous Clinical Evaluation Reports (CERs)
- Managing EUDAMED registration
- Aligning Quality Management Systems with Annex IX/X conformity assessment procedures
The Notified Body backlog compounds these challenges. As of October 2022, fewer than 2,000 MDR certificates had been issued against 22,793 valid MDD/AIMDD certificates — a gap that puts serious timeline pressure on new market entrants.
Navigating these requirements without prior EU regulatory experience makes specialist guidance critical. This article identifies the top 10 EU MDR consulting firms specifically equipped to guide US medical device companies through CE marking and ongoing EU compliance in 2026.
Key Takeaways
- EU MDR replaced the MDD with stricter clinical evidence, post-market surveillance, and documentation requirements — a heavier compliance burden for US manufacturers
- US companies must appoint an EU Authorized Representative and navigate complex Annex requirements; specialized consultants handle both
- Top consultants are selected based on EU regulatory track record, technical documentation depth, CER expertise, and proven experience with US-based manufacturers
- Leading firms include Elexes, Emergo by UL, Qserve Group, and RQM+, each offering distinct capabilities for US companies pursuing CE marking
- Selection depends on device class, timeline, budget, and whether you need comprehensive support or specialist intervention
What US Medical Device Companies Need to Know Before Choosing an EU MDR Consultant
EU MDR imposes specific requirements on foreign manufacturers that differ significantly from FDA regulations. Article 11 mandates that all non-EU manufacturers designate an EU Authorized Representative established in an EU member state.
Companies must also implement Unique Device Identification (UDI) systems, complete EUDAMED registration (mandatory by May 28, 2026), and conduct post-market clinical follow-up (PMCF) — none of which have direct FDA equivalents.
US companies typically need specialized support across several domains:
- Device classification under MDR Annex VIII (rules differ from FDA classification)
- Technical documentation preparation per Annex II/III requirements
- Clinical Evaluation Reports (CER) and PMCF plans meeting EU standards
- ISO 13485 QMS alignment with EU requirements
- Notified Body selection, application, and audit coordination
According to MedTech Europe's 2024 report, average MDR certification timelines have reached 19.5 months for QMS certification and 21.8 months for Technical Documentation Assessment. Complex Class IIb/III devices typically require 19–24 months total. The firms below were selected for documented EU MDR expertise and a track record supporting US manufacturers in the European market.
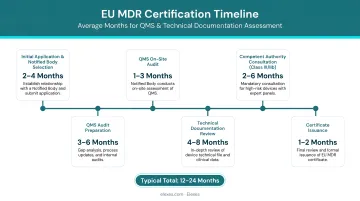
Top 10 EU MDR Consultants for US Medical Device Companies 2026
These consulting firms were evaluated based on EU MDR-specific expertise, documented CE marking submission outcomes, experience addressing US manufacturer requirements (EU Rep services, technical file preparation), team credentials (former Notified Body staff, EU regulatory backgrounds), and flexible engagement models.
Elexes
A globally recognized regulatory consulting firm with 50+ years of collective experience and 256+ global clients, Elexes delivers end-to-end EU MDR support tailored to US manufacturers—from device classification and technical file preparation through Notified Body coordination and post-market surveillance. The firm maintains a 90% audit clearance rate and has classified 200+ device types under EU MDR.
Flexible engagement models—full-time embedded, part-time, or project-based—let US companies scale regulatory support without permanent headcount expansion. The cross-functional team covers regulatory, quality, and clinical tasks across SaMD, IVDs, implants, wearables, and combination devices, with demonstrated dual-pathway capability for companies managing simultaneous FDA and EU MDR compliance.
| Key EU MDR Services | CE marking, CER/PMCF, ISO 13485 QMS, technical documentation, EUDAMED registration, post-market surveillance |
|---|---|
| Notable Strength | End-to-end EU MDR support with FDA dual-pathway capability; 50+ years collective experience |
| Best For | US startups to mid-size companies needing flexible, full-service EU MDR and FDA compliance support |
Emergo by UL
One of the most recognized global medical device consulting firms (acquired by UL in 2019), Emergo employs 400+ regulatory professionals with offices in the Netherlands, Ireland, UK, and Germany—strategically positioned to support US companies pursuing CE marking under EU MDR. This extensive European presence provides direct access to Notified Body networks and local regulatory expertise.
The breadth of services covers EU MDR/IVDR submissions, Authorized Representative services, ISO 13485 implementation, and multi-country registration in 40+ countries. Emergo's established Notified Body liaison network across Europe can reduce certification timelines, though prospective clients should note that UL's Notified Body designation creates potential conflicts for some engagements.
| Key EU MDR Services | CE marking, EU Authorized Representative, CER, ISO 13485, global registration, EUDAMED |
|---|---|
| Notable Strength | Largest global reach; strong Notified Body relationships and multi-country capability |
| Best For | Mid-to-large US manufacturers seeking single partner for EU MDR plus multi-country registration |
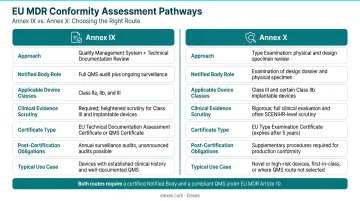
Qserve Group
A Netherlands-based specialist founded in 1998, Qserve Group employs consultants with direct Notified Body and industry backgrounds, providing insider understanding of EU MDR conformity assessment expectations. The firm has documented 750+ MDR submissions, demonstrating substantial hands-on experience with the current regulation.
Qserve excels in clinical evaluation depth, Annex IX/X conformity assessment guidance, and PMCF strategy—critical capabilities for US companies facing stringent clinical evidence requirements. The firm's exclusive EU MDR/IVDR focus (no competing service lines) ensures concentrated expertise, valuable for Class IIb/III devices and IVD manufacturers entering the European market.
| Key EU MDR Services | CER/PMCF, EU MDR technical files, Annex IX/X, ISO 13485, Notified Body liaison, IVD/IVDR |
|---|---|
| Notable Strength | Former Notified Body consultants with 750+ MDR submissions; IVD and diagnostics specialty |
| Best For | US companies needing deep EU MDR/IVDR expertise, especially Class IIb/III devices or IVDs |
RQM+
A pure-play life sciences regulatory and quality consulting firm with 400+ consultants, RQM+ was formed from multiple established consultancies and maintains dedicated EU MDR/IVDR practices across UK, Ireland, Germany, and Netherlands offices. The firm's European footprint provides local market knowledge combined with US accessibility.
Remediation is where RQM+ stands apart: former Notified Body professionals bring firsthand knowledge of audit expectations and deficiency resolution strategies, making the firm a strong choice for US companies already receiving nonconformities. Gap assessments and Technical Documentation reviews round out a tightly focused service offering.
| Key EU MDR Services | EU MDR gap assessment, Technical Documentation, CER, PMCF, ISO 13485, MDSAP, EU IVDR |
|---|---|
| Notable Strength | Nonconformity remediation led by former Notified Body professionals |
| Best For | US companies responding to EU MDR nonconformities or undergoing Notified Body audits |
Veranex
Formed through combination of several established consultancies with European headquarters in France, Veranex offers integrated product development and regulatory consulting with deep EU MDR capabilities across US, France, UK, Germany, and Ireland. The firm employs 800+ cross-disciplinary experts serving 700+ global clients.
What sets Veranex apart is the combination of product design and engineering services with EU MDR regulatory consulting. This integration benefits US companies developing new devices who want design-for-compliance built in from the start. The "Innovation CRO" model reduces the gap between engineering and regulatory functions, potentially accelerating time-to-market.
| Key EU MDR Services | CE marking, CER, design controls, ISO 14971 risk management, EU MDR technical files, clinical affairs |
|---|---|
| Notable Strength | Unique integration of product development and EU MDR regulatory consulting |
| Best For | US companies at development stage seeking EU MDR-ready design controls and regulatory strategy |
PharmaLex (part of Cencora)
A Germany-headquartered regulatory consulting firm with deep EU regulatory roots, PharmaLex is trusted by global medtech firms for EU MDR clinical evaluation, IVDR support, and ongoing post-market compliance—now backed by Cencora's (formerly AmerisourceBergen) global infrastructure following acquisition in January 2023.
European-native regulatory expertise and strong Clinical Evaluation Report (CER) writing capabilities make PharmaLex a natural fit for US companies facing stringent clinical evidence demands under EU MDR Class IIb and III requirements. Regulatory intelligence tracking helps clients stay ahead of evolving EU guidance.
| Key EU MDR Services | CER/PMCF, EU MDR strategy, IVDR compliance, post-market surveillance, QMS, regulatory intelligence |
|---|---|
| Notable Strength | European-native expertise; strong CER and clinical evidence depth for high-risk devices |
| Best For | US companies with Class IIb or Class III devices requiring high-quality clinical documentation |
NAMSA
A full-service CRO and consulting firm founded in 1967 with 1,700+ associates and offices in the US and Europe, NAMSA uniquely combines EU MDR regulatory consulting with in-house laboratory testing capabilities for biocompatibility and sterilization validation. This integrated model serves 3,000+ companies each year.
The consulting and testing integration lets US companies handle ISO 10993 biocompatibility evaluation, sterilization validation, and EU MDR technical documentation under one roof. This reduces coordination burden and eliminates timeline delays from managing multiple vendors.
| Key EU MDR Services | CE marking, CER, biocompatibility testing (ISO 10993), sterilization validation, clinical trial management |
|---|---|
| Notable Strength | Only firm combining EU MDR regulatory consulting with in-house biocompatibility and sterilization labs |
| Best For | US manufacturers of implantable or sterile devices needing integrated testing and regulatory support |
Freyr Solutions
An India- and US-headquartered regulatory consulting firm with services across 120+ countries, Freyr employs 2,500+ regulatory professionals and offers cost-effective EU MDR support including technical documentation, labeling/UDI, and regulatory lifecycle management through a scalable delivery model.
Competitive pricing without sacrificing global reach makes Freyr a practical choice for US companies with cost sensitivity who need efficient EU MDR technical file preparation, labeling compliance, and EUDAMED registration support. Regulatory intelligence tracking across EU and global markets helps clients navigate changing requirements.
| Key EU MDR Services | EU MDR technical files, labeling/UDI, EUDAMED, post-market surveillance, regulatory lifecycle management |
|---|---|
| Notable Strength | Cost-effective delivery model; strong regulatory intelligence tracking across global markets |
| Best For | Budget-conscious US companies with large device portfolios needing efficient documentation support |
MDI Consultants
A US-based medical device consulting firm with experience in both FDA and EU regulatory pathways, MDI Consultants helps US manufacturers navigate the dual compliance challenge of maintaining FDA compliance while pursuing CE marking. The firm has been active in medical device consulting since 1994.
US-based accessibility is MDI's core advantage. Clients work with a team that understands FDA QSR/QMSR requirements alongside EU MDR, reducing internal coordination barriers. Time zone alignment and cultural familiarity can meaningfully accelerate project execution for US teams.
| Key EU MDR Services | CE marking, EU MDR technical documentation, ISO 13485, clinical evaluation support, FDA-EU dual pathway |
|---|---|
| Notable Strength | US-based team with dual FDA and EU MDR expertise for companies managing both pathways simultaneously |
| Best For | US companies seeking integrated FDA compliance and EU MDR support from a single, US-accessible partner |
Regulatory Compliance Associates (RCA)
A life sciences regulatory and quality consulting firm with 100+ consultants and US headquarters, RCA supports US manufacturers with EU MDR compliance alongside FDA submissions, offering broad device category coverage and a strong remediation practice. The firm is part of the Nelson Labs/Sotera Health group.
FDA remediation experience is where RCA adds distinct value alongside EU MDR support. US companies that have received Warning Letters or 483 observations benefit from a systems-level compliance approach that addresses root causes rather than isolated deficiencies—useful context when also preparing for EU MDR certification.
| Key EU MDR Services | EU MDR compliance, CE marking, ISO 13485, CER support, QMS remediation, Notified Body audit preparation |
|---|---|
| Notable Strength | Strong compliance remediation practice; broad Class I–III device category coverage for US and EU |
| Best For | US companies with existing compliance gaps needing systematic QMS remediation before EU MDR certification |
How We Chose the Best EU MDR Consultants for US Companies
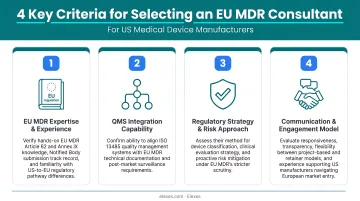
Each firm on this list was evaluated across four criteria:
- EU MDR track record — documented CE marking outcomes and Clinical Evaluation Report (CER) quality
- US foreign manufacturer experience — EU Authorized Representative services and Annex II/III technical documentation preparation
- Team credentials — former Notified Body staff, EU-native regulatory backgrounds
- Engagement flexibility — project-based, retainer, and embedded models suited to varying company sizes and budgets
US companies should prioritize several key factors when selecting an EU MDR consultant:
- EU Authorized Representative capability – Confirm the firm offers Article 11 EU AR services directly or through vetted partnerships.
- Notified Body liaison experience – Look for firms that have cleared audits across multiple Notified Bodies and maintain active working relationships with them.
- Device class and product type alignment – Verify documented submissions in your specific device classification and technology category — not just general MDR experience.
- Engagement model fit – Match their project-based, retainer, or embedded model to your timeline, internal bandwidth, and budget before committing.
Even with these criteria in hand, US companies still fall into predictable traps. The most common:
- Choosing FDA-focused firms that lack genuine EU MDR depth
- Underestimating the value of EU-native team members who know local regulatory culture firsthand
- Selecting based on brand name rather than verifiable EU MDR submission outcomes and references in comparable device categories
Conclusion
EU MDR compliance represents a substantial undertaking for US medical device companies entering or maintaining presence in the European market. The right consulting partner can mean the difference between a smooth CE marking process and costly nonconformities or Notified Body rejections that delay market access by months or even years. With average certification costs exceeding 136,000 EUR for QMS and 176,000 EUR for Technical Documentation Assessment, selecting a consultant with proven EU MDR expertise is a critical investment decision.
US companies should evaluate consultants on EU MDR-specific credentials, not just global reputation. Key criteria to assess:
- Number of Technical Documentation reviews completed under the current regulation
- CER writing capability with documented Notified Body acceptance rates
- Established Notified Body relationships that can facilitate direct communication
- Direct experience with US foreign manufacturer requirements under Article 11, including EU AR designation
Elexes supports US medical device companies through every stage of EU MDR compliance — device classification, technical file preparation, Notified Body coordination, and post-market surveillance — with flexible engagement models and 50+ years of collective regulatory experience across 256+ global clients. Whether you're a startup pursuing your first CE mark or an established manufacturer transitioning from MDD to MDR, our team brings EU-specific expertise with direct US accessibility. Contact us today to discuss your EU MDR project.
Frequently Asked Questions
What is EU MDR and why do US medical device companies need to comply with it?
EU MDR (Regulation (EU) 2017/745) is the European Union's current medical device regulation, which replaced the Medical Device Directive (MDD) in May 2021. Any US company selling devices in the EU/EEA market must comply. See Q6 below for a breakdown of how MDR differs from the previous MDD requirements.
Do US medical device companies need an EU Authorized Representative under EU MDR?
Yes, Article 11 of EU MDR requires all non-EU manufacturers to designate an EU Authorized Representative established in an EU member state before placing devices on the European market. Most EU MDR consulting firms either provide this service directly or connect you with an established EU AR partner — confirm this capability before engaging any consultant.
How long does EU MDR certification typically take for a US company?
Timelines depend on device class and Notified Body workload. Class IIa devices typically take 12-18 months; Class III devices can take 24-36 months or more. Start Notified Body engagement at least 12-18 months before your planned submission to account for ongoing capacity constraints.
What documents are required for EU MDR technical documentation for a US manufacturer?
Core EU MDR Annex II/III requirements include:
- Device description and specifications
- Design and manufacturing information
- EU Declaration of Conformity
- Risk management file (ISO 14971)
- Clinical Evaluation Report (CER)
- Post-market surveillance plan
- Labels and instructions for use
Each document must map to the applicable General Safety and Performance Requirements (GSPRs) with supporting evidence.
How much does EU MDR consulting typically cost for a US medical device company?
Costs vary widely based on device complexity and company readiness. Full CE marking projects for Class IIa devices may range from $40,000 to $120,000, while Class III projects can exceed $200,000. Consulting fees depend on device complexity, clinical data requirements, whether the QMS is already ISO 13485 certified, and the engagement model selected. Notified Body fees add substantially to total costs.
What is the difference between EU MDR and the previous Medical Device Directive (MDD)?
MDR significantly raised the bar compared to MDD across several areas:
- Mandatory CERs for all device classes (not just higher-risk)
- Mandatory PMCF studies for higher-risk devices
- EUDAMED registration for manufacturers and devices
- Structured PMS plans and Periodic Safety Update Reports (PSURs)
- Expanded UDI requirements
- More rigorous Notified Body designation and oversight
MDD-certified devices required recertification under MDR by applicable transition deadlines: December 2027 for Class III, December 2028 for lower classes.


