

Design History File (DHF) Remediation for Medical Devices – Process, Steps & Best Practices
Behind every innovation, there are years of hard work, passion, and perseverance. A medical device based on innovation, meticulous planning, and excellent execution can transform lives. To protect these innovations, we often focus on patents, legal agreements, and NDAs. However, many tend to overlook the importance of documenting the most critical element of innovation—the design itself. This is where DHF Remediation becomes important. As the product evolves, so must the design history file (DHF), ensuring that changes and decisions are appropriately documented to meet regulatory standards and stay compliant.
Elexes has helped medical device manufacturers remediate their DHFs post‑audit and maintain compliance with FDA and ISO 13485 requirements. As a QA/RA Director of a medical device firm, you know legacy device documentation poses audit risk – here’s how to remediate your DHF and stay inspection‑ready.
What is the Need for a DHF?
Section 7.3.10 in ISO 13485:2016 and 21 CFR part 820.30 permeates that ‘each manufacturer shall establish and maintain a DHF for each type of device. The Design History File (DHF) shall contain or reference the records necessary to demonstrate that the design was developed in accordance with the approved design plan and the requirements of this part’. The design of the product with its specifications, design review, verification and validation, and so on should be recorded in a Design History File (DHF). In simple words, a DHF is nothing but a compilation of design history records of a finished device.
Design and control process for any medical device

Once the design of the medical device is frozen, it is transferred to manufacturing. The design transfer indicates that the DHF is finalized. After the design transfer, any changes to any design specifications, or software updates must undergo a Change Control Process.
This process details the change, what is being affected, what inputs or outputs are affected, the risk, priority level, activities needed to implement the change such as regulatory approvals or verification/validation of design change, and a verification that the change has been implemented. Following this the DHF should be updated to reflect the change.
DHF Remediation an important but forgotten endeavor
It is evident that the DHF is typically a living file that requires constant updation throughout the design life cycle. This is where persistence plays a vital role for any medical device company, as it has to maintain constant compliance with the several regulatory design control requirements and regulations which are dynamic.
However, several years back neither was there a good understanding on how often and when the DHF should be updated, nor did the Auditors or Inspectors check frequently for updation; as a result, amidst the fast pace of sales, marketing, changing team members, shifting priorities, post-marketing surveillance, more often than not the DHF update was forgotten.
In such cases, or in cases of a significantly changed standard or regulation, DHF remediation is certainly called for to protect what device companies have innovated and to prevent any eventual market recall issues.
“Remediation – Easier said than done”
Remediating a DHF is a colossal endeavor that requires multiple resources’ time and effort to complete this detail-oriented task and companies often have to dedicate personnel from other functional teams for remediation, which is not very efficient or in the best interest of the company.
That’s where Elexes could step in and support these companies in conducting the remediation process for accurate updates to the DHF, charted on a predetermined timeline.
How should companies really go about implementing a DHF Remediation?
5 Steps To Consider While Implementing a DHF Remediation
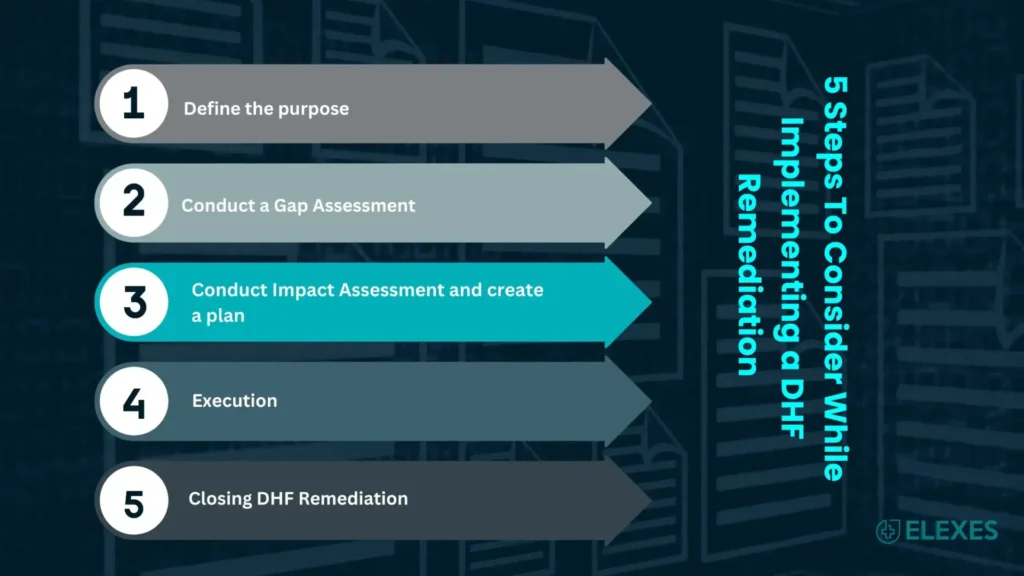
Step 1: Define the purpose
The purpose for DHF Remediation must be defined in order to lay down objective steps for the remediation. DHF could be remediated for:
Step 2: Conduct a Gap Assessment
This is an important step to understand the “delta” between what is the target (based on the purpose defined in step 1) and what exists now. This step will reveal all the gaps that need to be addressed for complete DHF remediation.
Step 3: Conduct Impact Assessment and create a plan
Once the gap assessment is complete, the impact of the each of the “delta” must be evaluated, e.g., if re-testing needs to be done, if risk assessment needs to be updated and what kind of update (creating a risk-benefit analysis, or adding more hazards based on product’s field experience). This impact assessment would help enlist action items based on which a plan that would be elementary to the successful execution would be defined. A plan should contain:
Who? People and the kind of expertise, background and skill set that should be involved
How Many? Number of personnel to be involved both for planning and for execution
Tools? Checklists, cloud-based platforms to collaborate, or document repository
Time? Overall time required
Step 4: Execution
This is the most time-consuming step and involves the implementation of the plan to the most minute action items. Successful execution requires a strong team who is committed to achieving the end goal.
Step 5: Closing DHF Remediation
This is the last step where everyone signs off on the revised DHF and it forms a solid base for the product for many years to come.
For medical device manufacturers, an organized DHF is ideal for laying a good foundation of the quality system through the design and development, verification and validation testing, clinical trials, and post-market phases. The indispensable need to align the strategies adopted with regulatory requirements is perhaps discernible.
Let’s protect our innovations of the past with equal diligence with which we protect the new ones. For any questions/comments please write to connect@elexes.com
Talk to a DHF Remediation Expert for Medical Devices
FAQs
What is DHF remediation and when is it needed for medical devices?
DHF remediation is the process of updating an existing Design History File to meet current regulatory and quality standards; it’s needed when gaps are identified during audits, regulatory submissions, or legacy product reviews.
What are the regulatory requirements for a Design History File (DHF) under FDA and ISO?
A DHF must demonstrate compliance with design control requirements under FDA 21 CFR 820.30 and ISO 13485, including documented design planning, inputs, outputs, verification, validation, and design changes.
What are common gaps found in DHFs of legacy medical devices?
Common gaps include missing verification and validation documentation, incomplete traceability, undocumented design decisions, and inadequate change control records.
How long does DHF remediation take and what resources are needed?
Remediation timelines vary from weeks to months depending on complexity of the device; it typically needs cross-functional SME’s, quality engineers, and document control support.
How can DHF remediation prevent audit findings and recalls?
DHF remediation ensures documented evidence of design controls and reduces non-conformances, thereby lowering the risk of audit observations and market recalls.



















